
Abstract
Mixed Connective Tissue Disease (MCTD) is an autoimmune disease first described by Sharp et al in 1972, characterized by the presence of anti-Ribonucleoprotein antibodies directed against the U1 complex (anti-U1RNP). The condition shares clinical characteristics with Systemic Lupus Erythematosus, Rheumatoid Arthritis, and Systemic Sclerosis. Diagnosis is quite difficult due to its rarity, the lack of validated classification criteria, and its heterogeneous clinical presentation. While in the early stages its nuanced clinical features might lead to it being incorrectly classified as other Connective Tissue Diseases (CTDs) or even not recognized, in cases of longstanding disease its classification as a CTD is clear but challenging to discriminate from overlap syndromes. MCTD should be considered a distinct entity due to the presence of a specific genetic substrate and the presence of the high titer of a specific autoantibody, anti-U1RNP, present in all the commercial kits for Extractable Nuclear Antigens, and almost always associated with Antinuclear Antibody positivity with a coarse speckled pattern. Except for anti-U1RNP, no specific biomarkers are available to guide clinicians to a correct classification of MCTD, which is arrived at by the association of clinical, serological and instrumental evaluation. In the first stages, the disease is mainly characterized by Raynaud’s phenomenon, inflammatory arthritis, puffy fingers, myalgia and/or myositis, and rarely, trigeminal neuropathy. Longstanding disease is generally associated with the development of Pulmonary Hypertension and Interstitial Lung Disease, which are the two main causes of mortality in MCTD. The aim of this review is to summarize current knowledge on the early recognition of MCTD.
Introduction
The term “Mixed Connective Tissue Disease” (MCTD) refers to a rare autoimmune disease, first described by Sharp et al in 1972, that shares clinical features with Polymyositis (PM), Systemic Sclerosis (SSc), Systemic Lupus Erythematosus (SLE), and Rheumatoid Arthritis (RA).Citation1 Despite its heterogeneous clinical presentation, it is generally accepted that MCTD should be considered a distinct clinical entity rather than an overlap condition, being associated with a specific autoantibody: anti-U1-Ribonucleoprotein (anti-U1RNP).Citation1
The epidemiology of MCTD is affected by several potential biases, due to its rarity, heterogeneous presentation, and the presence of multiple subsets of proposed classification criteria to define the condition.Citation2–6 Referring to the two studies that included patients enrolled according to the four subsets of classification criteria available at the time of the study,Citation7,Citation8 the incidence of MCTD is reported at 2.1–19 per million per year, with a prevalence of about 3 per 100,000 persons, and a female-to-male ratio of 3–5:1. Despite its multi-organ involvement, mortality was described as similar to the general population.Citation7
The pathophysiology of this condition is also largely unknown, but it clearly seems to be multifactorial. A genetic association with the development of MCTD was found in patients with HLA-DR4, but not with HLA-DR3 and -DR5 (which are strongly associated with SLE and SSc respectively).Citation9 This data could further support the definition of MCTD as a distinct entity. Environmental and occupational exposure in predisposed patients, as well as infections (mainly retroviruses such as Human Immunodeficiency Virus Type-1) seem to play a critical role in the development of the condition.Citation10,Citation11 The hallmark of the disease is represented by anti-U1RNP (or anti-nRNP),Citation12 an autoantibody directed against the 70kDa subunit of the U1 RNP complex, an intracellular protein involved in mRNA maturation.Citation13 It is possible that during the process of apoptosis, a modified U1 70kDA protein is exposed on the surface of the apoptotic blebs, which is then recognized by antigen-presenting cells, presented to T cells, leading ultimately to B cell stimulation.Citation14 Anti-U1-RNP can stimulate both mononuclear cells and autoreactive CD4+ cells, with an increased production of Interleukin (IL) 1 and IL6 (leading to autoimmune inflammation), but tissue injury is also sustained due to the direct action of the autoantibody on endothelial cells. It is able to induce the production of intercellular and endothelial leukocyte adhesion molecule-1 (ICAM-1 and ELAM-1, respectively) which also act as anti-endothelial cells.Citation14 This mechanism could explain the vascular involvement seen in MCTD, in particular Pulmonary Artery Hypertension (PAH). The presence of double stranded RNA in the U1 complex is responsible for the activation of innate immunity through Toll-like Receptor 3, activating also the innate immunity.Citation14 Anti-U1RNP also seems to be able to activate immature dendritic cells via TLR7, inducing the production of Type I Interferon (IFN-1), in turn able to promote the activation of autoreactive T cells, with a down-regulation of regulatory T cells and the production of effector T cells, ultimately responsible of the tissue injury. IFN-1 also activates autoreactive T cells, leading to the production of autoantibodies.Citation15 Anti-U1RNP seems also have a prognostic value: high titre of anti-U1RNP seems to be associated with the presence of PAH and a less common neurological involvement, whereas its disappearance is associated with a persistent disease remission.Citation15–17
Clinical Presentation and Classification Criteria
The initial clinical picture of MCTD patients is generally characterized by non-specific symptoms such as low-grade fever, arthralgia and fatigue.
Generally, the first specific involvement includes the skin. MCTD is potentially associated with a skin involvement common in SLE and above all SSc, therefore it is almost impossible to distinguish between these two conditions through a simple clinical evaluation. The first symptom is generally Raynaud’s Phenomenon (RP). During follow-up, any of the typical clinical features of SSc can occur, from puffy hands to sclerodactyly, acrosclerosis, calcinosis, ulcers and digital necrosis.Citation18 RP is present in almost all MCTD patients from the first assessment onwards, whereas fibrotic changes of the skin became more common during follow-up, reaching a prevalence of about 90%.Citation19 Oral ulcers, discoid lesions, panniculitis and the classical SLE malar rash are also common in MCTD.Citation19
Inflammatory arthritis is also a common clinical feature in MCTD, generally present from the first stage of the disease onward. Joint involvement resembles classic RA, and positivity for Rheumatoid Factor (RF) and Anti Citrullinated Protein Antibody (ACPA) are noted in 70% and 50% of patients respectively.Citation20 This inflammatory arthritis is potentially erosive, potentially leading to the typical deformities associated with RA.Citation21
Inflammatory myopathy is also very common and is necessary, under all of the proposed criteria, for classification as MCTD. Also in this case, MCTD might be clinically indistinguishable from an idiopathic inflammatory myopathy, with proximal weakness, typical alteration at electromyography and increased serum level of muscle enzymes such as Creatine Phosphokinase (CPK), Lactic Dehydrogenase (LDH), transaminases.Citation22 Muscle samples from MCTD patients show muscle cellular injury similar to that shown in PM patients, together with the vascular changes reported in Dermatomyositis patients.Citation23 It should be noted that most patients only report diffuse myalgia, which could be the expression of mild, silent, or localized myositis.Citation24
From the gastrointestinal point of view, MCTD shows involvement that is common in SSc, SLE and PM. Gastroesophageal Reflux with heartburn and dysphagia is present in about 50% of patients, and is the most common feature.Citation25 Almost any part of the gastrointestinal tract could be involved during MCTD, although generally it is very uncommon. Peristalsis disturbance, hepatitis, pancreatitis, malabsorption and small bowel perforation secondary to vasculitis are also possible.Citation26
The cardiovascular system is also commonly involved. Pericarditis is present, though often subclinically, in about 40% of patients, while heart rhythm disturbances affect about 20%.Citation27 A large proportion of patients show intimal proliferation and medial hypertrophy of medium- and small-sized vessels.Citation28 This mechanism, characteristic of the peripheral vasculopathy responsible for ulcers in the fingers, is potentially also present in the pulmonary vasculature, leading to PAH. PAH is present in about 20% of MCTD patients and represents one of the main causes of mortality.Citation29 Other potential mechanisms associated with PH are chronic embolism and Interstitial Lung Disease (ILD).Citation30
Lung involvement is also very common. The most important condition in terms of prevalence and prognostic impact is ILD, affecting about 60% of MCTD patients.Citation31 Patients display features quite similar to those reported in SSc: High-Resolution Computed Tomography (HRCT) shows the prevalence of a fibrotic Nonspecific Interstitial Pneumonia (NSIP) pattern (in nearly 90% of patients), whereas about 10% of patients have a definite Usual Interstitial Pneumonia (UIP) pattern.Citation32
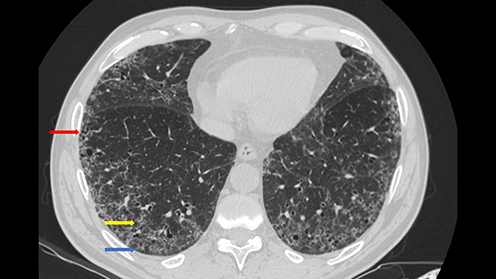
shows a typical fibrotic NSIP pattern in a patient with longstanding MCTD.
Figure 1 Fibrotic Nonspecific Interstitial Pneumonia in a patient with a longstanding Mixed Connective Tissue Disease. The pattern is characterized by diffuse Ground Glass Opacities (yellow arrow), subpleural sparing (blue arrow), and presence of traction bronchiectasis (red arrow).
Pleurisy is also reported in about 50% of patients, and it can be a rarely presenting manifestation of MCTD, but is generally mild and does not require specific treatment.Citation33 Rare and severe manifestations are, as reported for SLE: pulmonary vasculitis, pulmonary hemorrhage and thromboembolism associated with Anti-Phospholipid Syndrome (APS).Citation33
From the neurological point of view, as reported for SSc, the most common presentation is trigeminal neuropathy. It is present in about a quarter of patients, is a possible clinical presentation of MCTD, and was included in the last version of proposed classification criteria.Citation6,Citation34,Citation35 Other manifestations are generally mild, characterized by headaches, cognitive impairment, depression, and hearing loss. Similarly to SLE, psychosis, seizure and encephalitis are possible.Citation34
MCTD patients may also experience kidney damage. It is present in about 25% of patients, mainly characterized by membranous nephropathy, but is generally mild.Citation36 In fact, anti-RNP could have a protective role against kidney involvement in both MCTD and SLE, irrespective from the titre.Citation37 Other kidney manifestations, typical of SLE and SSc (including scleroderma renal crisis) are also reported.Citation36
Finally, MCTD patients may also experience hematologic manifestations. The most common are hemolytic anemia, autoimmune thrombocytopenia and leukopenia, as also reported for SLE.Citation19 Anticardiolipin antibodies can be also present and they could be associated with APS, whereas some case reports describe thrombotic thrombocytopenic purpura.Citation19
The combination of these clinical findings allows for a classification as MCTD. As things currently stand, the American College of Rheumatology and the European League Against Rheumatism, the most important societies in rheumatology, have not yet provided a specific classification of MCTD. Of the four historical classification criteria,Citation2–5 the maximum sensitivity was shown by the Kasukawa criteria (77.5%) whereas the best specificity was found for the sets proposed by Alarcón-Segovia-Villareal and Kahn (both 99.4%).Citation38 In addition, a relatively recent new set of diagnostic criteria was proposed in 2019.Citation6
All of these criteria are reported in .
Table 1 Proposed Classification Criteria for Mixed Connective Tissue Disease
Early Diagnosis of MCTD
MCTD is a rare condition, and diagnosis is further complicated by the presence of overlapping features with other conditions. Distinguishing MCTD from other systemic rheumatic diseases, including overlap syndromes, is a clinical challenge due to the heterogeneity of disease presentations; misdiagnosis at presentation of the disease is thus common in up to 61% of a single center cohort. Undifferentiated CTD, non-MCTD overlap syndromes and SLE were the most frequent diagnoses mistaken for MCTD that were reported.Citation39 Some cases of MCTD have been shown to evolve over time into other systemic rheumatic diseases,Citation40 while other studies have reported that true phenotypic conversion is rare.Citation39
In the first stage of the disease, the challenge for physicians is to consider the presence of MCTD in patients with nuanced clinical features. Classical clinical presentation at onset includes RP, inflammatory arthritis, puffy fingers and, rarely, trigeminal neuropathy. These parameters, when present simultaneously, even if not sufficient to reach specific classification criteria, clearly suggest an autoimmune origin. However, when only inflammatory arthritis is present, inaccurate classification is possible, whereas in cases of an isolated onset of RP, clinicians should also consider the fact that this sign is also prevalent in the general population.
In advanced stages, the clinical picture unquestionably indicates the presence of a Connective Tissue Disease (CTD), however the difficultly lies in determining which cases are actually MCTD and not another specific condition such as SLE, SSc, or commonly, an overlap condition.
As already reported, visceral involvement of MCTD is frequently mild, and often asymptomatic, at least until the damage become clinically significant. Therefore, an appropriate diagnosis requires a systematic clinical, serological and instrumental approach.
Clinical Evaluation
Clinical evaluation can allow for the objectivation of the presence of RP and/or inflammatory arthritis. The presence of puffy fingers or sclerodactyly, together with the possible presence of skin lesions can allow for the recognition of a scleroderma-phenotype, thus directing diagnostic assessment. MCTD should be considered to be a disease included among the scleroderma spectrum disorders, therefore chest auscultation can allow for the recognition of Velcro crackles typically associated with ILD. They can be present in patients without typical symptoms such as dry cough and dyspnea, therefore chest auscultation could be of great interest in clinical practice.Citation41 An SLE-like phenotype of MCTD could be associated with malar rash, as well as decreased sound on chest auscultation, potentially associated with pleurisy. Patients could also display myalgia and proximal weakness, typically associated with a myositis phenotype.
However, clinical assessment alone is not sufficient, given that autoimmune assessment is fundamental to the recognition of anti-RNP, the hallmark of the disease, and the recognition of other potential markers associated with other conditions.
General Laboratory Evaluation
From laboratory evaluation, a complete blood count could reveal the presence of leukopenia and thrombocytopenia (both included in the sets of criteria created by Kasukawa and TanakaCitation4,Citation6), as well as the presence of hemolytic anemia. Increased levels of transaminases are possible, however, rather than being associated with hepatitis, which is very uncommon, they could be associated with myositis.Citation32 In this case, other specific parameters are LDH (also associated with lung damageCitation32) and CPK, levels of which are raised in about 30% of MCTD patients.Citation39 The increased serum level of CPK is quite widely accepted as a potential criterion for MCTD classification. Hypocomplementemia and presence of proteins in the urine can also be present (42% and 10% respectively) and support an early diagnosis.Citation39 Hypergammaglobulinemia is present in about 75%, and is generally polyclonal, however a peak can also be noted: in some cases, the anti-RNP titer represents about 20% of the total amount of immunoglobulins, thus mimicking a peak.Citation42
Autoimmune Assessment
Antinuclear Antibodies (ANA) are almost always present in MCTD, a with high titer showing a coarse speckled pattern.Citation39,Citation43 The presence of anti-U1RNP is required in order for the potential presence of a MCTD to be suspected, however the antibody could be also present in SLE and idiopathic inflammatory myopathies, so the full clinical picture should be considered. The antibody is included in the most common commercial kits for the assessment of Extractable Nuclear Antigens and currently represents the sole specific serological marker of MCTD. In the majority of patients, autoantibody positivity precedes the clinical onset of the disease, therefore initial assessment with ANA and ENA should be performed on all patients with suspected MCTD.Citation44 The presence of anti-double-stranded DNA (anti-DsDNA) is reported in about 30% of patients.Citation38 Episodic positivity for this autoantibody is possible in MCTD patients, however it should be borne in mind that this autoantibody has high specificity for active SLE, so the presence of an actual MCTD should be called into question in this case. Similar considerations can be made for ACPA, which is described in up to 50% of MCTD patients, but highly specific for RA.Citation20 The presence of anti-Ro60kDa, anti-Ro52kDa, and anti-La is reported in about 10% of patients.Citation38 Anti-La and, above all, anti-Ro60kDa are specific for Sjӧgren’s Syndrome (SS) and often associated with a significant case of sicca syndrome, allowing for the classification of MCTD patients as having secondary SS in up to 30% of cases.Citation45 Anti-Ro52kDa can be positive in a wide spectrum of autoimmune diseases, generally associated with myositis and ILD, therefore cases of positivity for this autoantibody should allow for appropriate assessment in patients.Citation32 Other autoimmune parameters commonly present in MCTD are direct Coombs test, present in about up to 50% of patients and potentially associated with hemolytic anemia, and lupus anticoagulant and/or anticardiolipin antibodies, present in about 10% of patients and potentially associated with APS.Citation38 These latter parameters clearly reflect a SLE phenotype and should raise questions about the presence of an actual MCTD or SLE.
Instrumental Evaluation
As RP is one of the most common clinical signs of early MCTD, Nailfold Videocapillaroscopy (NVC) is a cheap, rapid and useful tool for the assessment of the condition. A definite scleroderma pattern was found in 45–65% of cases at first assessment.Citation46,Citation47 At the early stages, the NVC pattern shows predominantly Giant Capillaries and neoangiogenesis, describing an early or active scleroderma pattern. In cases of longstanding disease, and mainly when associated with ILD, there is a predominance of avascular areas, and NVC shows a scleroderma pattern in all PAH-MCTD patients.Citation46–48 Interestingly, two different groups identified the possibility that immunosuppressive treatment is able to reverse NVC damage in MCTD, but not in SSc.Citation48,Citation49 This fascinating topic should be studied in depth in the coming years. Despite a strong association, NVC findings are not included in any set of MCTD criteria, however the exam is very useful in the assessment of RP as primary or secondary to a condition on the scleroderma spectrum disorder and can meaningfully guide diagnosis. The presence of a late pattern should also highlight the possibility of a high risk for digital ulcers and the presence of ILD. Finally, the presence of exaggerated neoangiogenesis could suggest the presence of an underlying associated myopathic phenotype.Citation49,Citation50
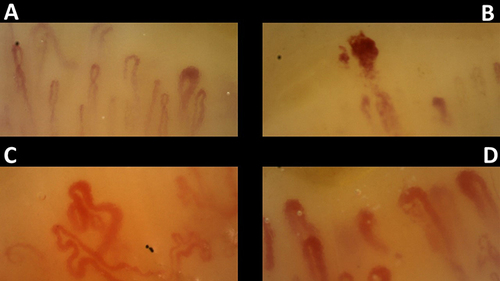
shows an NVC pattern from a female patient with MCTD.
Figure 2 Nailfold Videocapillaroscopy in a healthy subject (A) compared with a patient with Mixed Connective Tissue Disease (B–D). The pattern is characterized by the presence of multiple Giant Capillaries and neoangiogenesis.
Pulmonary function tests (PFTs) are highly useful in the early assessment of MCTD patients. Total Lung Capacity (TLC) and Forced Vital Capacity (FVC) are usually impaired in clinically significant ILD.Citation41 At the first assessment, abnormal PFTs are present in about a half of patients, with a slightly reduced FVC that remains stable after a decade.Citation51 On the contrary, Diffusing Lung Capacity for Carbon Monoxide (DLCO) decreases by about 15% during follow-up, showing a correlation with the worsening of the HRCT findings.Citation52 Despite the stability of FVC, about a quarter of patients showed a progressive-fibrosing phenotype of ILD, with a poor prognosis.Citation52,Citation53 These data could suggest DLCO as a sensitive parameter for the recognition of ILD and its worsening in CTD patients in general, and MCTD in particular. However, DLCO is not specific for the presence of ILD, as it is potentially also impaired in the presence of PAH.Citation41 A disproportionate relationship between FVC and DLCO values could suggest the presence of PAH. In SSc, a cut-off of 1.6 was proposed in the ratio between FVC and DLCO (both as proportion of predicted) as a clue for a possible underlying PAH.Citation41
PFTs are clearly useful for the initial assessment of patients, but on their own, are not sufficient for an appropriate assessment of the presence of ILD. As mortality in MCTD patients is mainly due to PAH or ILD, HRCT should be performed on all patients with a significant suspicion of MCTD. As already reported, the prevalent HRCT pattern is fibrotic NSIP, followed by UIP, although episodically consolidations as features of organizing pneumonia can also be seen, suggesting a myositis phenotype in the disease.Citation32 Studies on ILD associated with undifferentiated CTD have not proved an evolution towards MCTD, thus further supporting the idea that ILD is present in longstanding disease, or at least when the clinical picture is sufficient to reach a specific classification as MCTD.Citation54–56 HRCT can be also useful in evaluating possible pulmonary artery enlargement, as a clue to a possible, underlying PAH, which is relatively common in ILD-MCTD patients.Citation57
Appropriate screening for PAH can be carried out with a transthoracic echocardiogram, which should be considered a first-line exam in the assessment of MCTD patients. Subclinical cardiac abnormalities are present in up to 40% of patients.Citation58 The most common is pericardial effusion, and is useful in supporting an MCTD diagnosis, but the exam is also useful as screening for possible mitral valve prolapse, diastolic dysfunction, accelerated atherosclerosis and, above all, the possible presence of PAH.Citation27,Citation58 Despite the fact that a definite diagnosis of PAH can be made only after cardiac catheterization, transthoracic echocardiogram is useful in raising suspicion of the presence of the condition, and also in follow-up for monitoring response to treatment.Citation59
Conclusions
MCTD is a rare condition with heterogeneous presentations, which are potentially shared with all of the other CTDs. The correct classification of these patients can be difficult due to the absence of validated classification criteria, and nuanced clinical features at the onset. In this phase, the clinical picture could resemble more common CTDs, however without sufficient features for a specific classification, and patients could be classified as Undifferentiated Connective Tissue Diseases (UCTDs). On the other hand, in longstanding disease the presence of a specific CTD is easily recognized, but the patients could satisfy the classification criteria for at least two CTDs, therefore with a potential classification as overlap syndromes. Despite the fact that, conceptually, MCTD can be considered to be an overlap disease, its classification as a distinct entity is generally accepted. The condition is actually associated with the positivity of a specific autoantibody and a genetic predisposition associated with HLA-DR4 (whereas SLE and SSc are associated with HLA DR3 and DR5 respectively).Citation11 Moreover, despite a significant proportion of MCTD patients actually develop other signs able to satisfy also the classification criteria of other CTDs, the majority of patients remains stable with a specific clinical picture including RP, inflammatory arthritis and swollen hands.Citation40
An early diagnosis of MCTD generally starts with the recognition of RA-like inflammatory arthritis, RP, myalgia/myositis, and rarely, trigeminal neuropathy. These signs can be present alone or in combination. A correct early diagnosis can be made starting with the presence of the anti-U1RNP antibody, which is already present in patients before clinical disease onset. To correct classify patients, serological parameters such as complete blood count, hypocomplementemia, increased level of muscle enzymes and electrophoresis can support a suspicion of MCTD. NVC, PFTs, transthoracic echocardiogram and HRCT should be performed on all patients with a significant suspicion of MCTD.
The need for widely accepted and uniform criteria for MCTD diagnosis is required to allow further advances in knowledge on this topic. The last version of classification criteria is promising, however requires to be validated also in non-Asian Cohort.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
Gianluca Sambataro received honoraria from Boheringer Ingelheim, outside of the submitted work. Gaetano La Rocca, Chiara Alfia Ferrara, Giuseppe Ielo, and Alessandro Libra have no conflicts of interest to declare for this work.
Additional information
Funding
References
- Sharp GC, Irvin WS, Tan EM, Gould RG, Holman HR. Mixed connective tissue disease—an apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen (ENA). Am J Med. 1972;52(2):149–159. doi:10.1016/0002-9343(72)90064-2
- Sharp GC. Diagnostic criteria for classification of MCTD. In: Kasukawa R, Sharp GC, editors. Mixed Connective Tissue Disease and Anti-Nuclear Antibodies. Amsterdam: Elsevier Science Publishers B.V. (Biomedical Division); 1987:23–30.
- Alarcón-Segovia D, Villarreal M. Classification and diagnostic criteria for mixed connective tissue disease. In: Kasukawa R, Sharp GC, editors. Mixed Connective Tissue Disease and Antinuclear Antibodies. Amsterdam: Elsevier Science Publishers B.V. (Biomedical Division); 1987:33–40.
- Kasukawa R, Tojo T, Miyawaki S. Preliminary diagnostic criteria for classification of mixed connective tissue disease. In: Kasukawa R, Sharp GC, editors. Mixed Connective Tissue Disease and Anti-Nuclear Antibodies. Amsterdam: Elsevier Science Publishers B.V. (Biomedical Division); 1987:41–47.
- Kahn MF, Appelboom T. Syndrome de Sharp. In: Kahn MF, Peltier AP, Mayer O, Piette JC, editors. Les maladies systémiques. 3rd ed. Paris: Flammarion; 1991:545–556.
- Tanaka Y, Kuwana M, Fujii T, et al. 2019 Diagnostic criteria for mixed connective tissue disease (MCTD): from the Japan Research Committee of the Ministry of Health, Labor, and Welfare for systemic autoimmune disease. Mod Rheumatol. 2021;31(1):29–33. doi:10.1080/14397595.2019.1709944
- Ungprasert P, Crowson CS, Chowdhary VR, Ernste FC, Moder KG, Matteson EL. Epidemiology of mixed connective tissue disease, 1985–2014: a population-based study. Arthritis Care Rex. 2016;68(12):1843–1848. doi:10.1002/acr.22872
- Gunnarson R, Molberg O, Gilboe IM, Gran JT. The prevalence and incidence of mixed connective tissue disease: a national multicentre survey of Norwegian patients. Ann Rheum Dis. 2011;70(6):1047–1051. doi:10.1136/ard.2010.143792
- Hoffman RW, Rettenmaier LJ, Takeda Y, et al. Human autoantibodies against the 70-kD polypeptide of U1 small nuclear RNP are associated with HLA-Dr4 among connective tissue disease patients. Arthr Rheum. 1990;33:666–673. doi:10.1002/art.1780330509
- Zhao N, Smargiassi A, Chen H, Widdifield J, Bernatsky S. Systemic autoimmune rheumatic diseases and multiple industrial air pollutant emissions: a large general population Canadian cohort analysis. Environ Int. 2023;174:107920. doi:10.1016/j.envint.2023.107920
- Prokop J, Jagodzinski PP. Identification of retroviral conserved pol sequences in serum of mixed connective tissue disease and systemic sclerosis patients. Biomed Pharmacother. 2004;58(1):61–64. doi:10.1016/j.biopha.2003.10.001
- Lerner MR, Steitz JA. Antibodies to small nuclear RNAs complexed with proteins are produced by patients with systemic lupus erythematosus. Proc Natl Acad Sci. 1979;76(11):5495–5499. doi:10.1073/pnas.76.11.5495
- Greidinger EL, Zang YJ, Jaimes K, Martinez L, Nassiri M, Hoffman RW. CD4+ T cells target epitopes residing within the RNA-binding domain of the U1-70-kDa small nuclear ribonucleoprotein autoantigen and have restricted TCR diversity in an HLA-Dr4-transgenic murine model of mixed connective tissue disease. J Immunol. 2008;180(12):8444–8454. doi:10.4049/jimmunol.180.12.8444
- Keith MP, Moratz C, Tsokos GC. Anti-RNP immunity: implication for tissue injury and the pathogenesis of connective tissue disease. Autoimmun Rev. 2007;6(4):232–236. doi:10.1016/j.autrev.2006.08.007
- Paradowska-Gorycka A. U1-RNP and TLR receptors in the pathogenesis of mixed connective tissue disease. Part I. The U1-RNP complex and its biological significance in the pathogenesis of mixed connective tissue disease. Reumatologia. 2015;53(3):94–100. doi:10.5114/reum.2015.51509
- Hof D, Cheung K, De Rooij D, et al. Autoantibodies specific for apoptotic U1-70K are superior serological markers for mixed connective tissue disease. Arthritis Res Ther. 2005;7(2):R302–9. doi:10.1186/ar1490
- Xiang W, Dong R, Li M, Liu B, Ma Z, Yang Q. The role of anti-U1 RNP antibody in connective tissue disease-associated pulmonary artery hypertension: a systematic review and meta-analysis. J Clin Med. 2023;12(1):13. doi:10.3390/jcm12010013
- Sen S, Sinhamahapatra P, Chudhuri S, et al. Cutaneous manifestations of mixed connective tissue disease: study from a tertiary care hospital in eastern India. Indian J Dermatol. 2014;59(1):35–40. doi:10.4103/0019-5154.123491
- Pope JE. Other manifestations of mixed connective tissue disease. Rheum Dis Clin North Am. 2005;31(3):519–533. doi:10.1016/j.rdc.2005.04.011
- Takasaki Y, Yamanaka K, Takasaki C, et al. Anticyclic citrullinated peptide antibodies in patients with mixed connective tissue disease. Mod Rheumatol. 2004;14(5):367. doi:10.3109/s10165-004-0325-2
- Ramos-Niembro F, Alarcon-Segovia D, Hernandez-Ortiz J. Articular manifestation of mixed connective tissue disease. Arthritis Rheum. 1979;22(1):43. doi:10.1002/art.1780220107
- Hall S, Hanrahan P. Muscle involvement in mixed connective tissue disease. Rheum Dis Clin North Am. 2005;31(2):509. doi:10.1016/j.rdc.2005.04.003
- Vianna MA, Borges CT, Borba EF, Caleiro MT, Bonfà E, Marie SK. Myositis in mixed connective tissue disease: a unique syndrome characterized by immunohistopathologic elements of both polymyositis and dermatomyositis. Arq neuropsiquiatr. 2004;62(4):923. doi:10.1590/S0004-282X2004000600001
- Sambataro G, Ferrara CA, Spadaro C, et al. A new method for the assessment of myalgia in interstitial lung disease: association with positivity for myositis-specific and myositis-associated antibodies. Diagnostics. 2022;12(5):1139. doi:10.3390/diagnostics12051139
- Nica AE, Alexa LM, Ionescu AO, Andronic O, Padararu DN. Esophageal disorders in mixed connective tissue disease. J Med Life. 2016;9(2):141–143.
- Marshall JB, Kretschmar JM, Gerhardt DC, et al. gastrointestinal manifestations of mixed connective tissue disease. Gastroenterology. 1990;98(5 pt 1):1232. doi:10.1016/S0016-5085(12)90338-8
- Ungprasert P, Wannatong T, Panichsillapakit T, et al. Cardiac involvement in mixed connective tissue disease: a systematic review. Int J Cardiol. 2014;171(3):326–330. doi:10.1016/j.ijcard.2013.12.079
- Alpert MA, Goldberg SH, Singsen BH, et al. cardiovascular manifestations of mixed connective tissue disease in adults. Circulation. 1983;68(6):1182. doi:10.1161/01.CIR.68.6.1182
- Chaigne B, Chevalier K, Boucly A, et al. In-depth characterization of pulmonary arterial hypertension in mixed connective tissue disease: a French national multicenter study. Rheumatology. 2023. doi:10.1093/rheumatology/kead055
- Fayed H, Coghlan JG. Pulmonary hypertension associated with connective tissue disease. Semin Respir Crit Care med. 2019;40(2):173–183. doi:10.1055/s-0039-1685214
- Jeganathan N, Sathananthan M. Connective tissue disease-related interstitial lung disease: prevalence, patterns, predictors, prognosis, and treatment. Lung. 2020;198(5):735. doi:10.1007/s00408-020-00383-w
- Sambataro D, Sambataro G, Pignataro F, et al. Patients with interstitial lung disease secondary to autoimmune diseases: how to recognize them? Diagnostics. 2020;10(4):208. doi:10.3390/diagnostics10040208
- Prakash UBS. Respiratory complications in mixed connective tissue disease. Clin Chest Med. 1998;19(4):733–746. doi:10.1016/S0272-5231(05)70113-1
- Bennett RM, Bong DM, Spargo BH. Neuropsychiatric problems in mixed connective tissue disease. Am J Med. 1978;65(6):955. doi:10.1016/0002-9343(78)90747-7
- Hojaili B, Barland P. Trigeminal neuralgia as the first manifestation of mixed connective tissue disorder. J Clin Rheumatol. 2006;12(3):145. doi:10.1097/01.rhu.0000222045.70861.a5
- Lundberg IE. The prognosis of mixed connective tissue disease. Rheum Dis Clin North Am. 2005;31(3):535. doi:10.1016/j.rdc.2005.04.005
- Kitridou RC, Akmal M, Turkel SB, Ehresmann GR, Quismorio FB, Massry SG. Renal involvement in mixed connective tissue disease: a longitudinal clinicopathologic study. Semin Arthritis Rheum. 1986;16(2):135. doi:10.1016/0049-0172(86)90047-8
- John KJ, Sadiq M, George T, et al. Clinical and immunological profile of mixed connective tissue disease and a comparison of four diagnostic criteria. Int J Rheumatol. 2020;2020:9692030. doi:10.1155/2020/9692030
- Wanzenried A, Garaiman A, Jordan S, Distler O, Maurer B. the enigma of mixed connective tissue disease- challenges in routine care. Clin Rheumatol. 2022;41(11):3503–3511. doi:10.1007/s10067-022-06286-w
- Cappelli S, Bellando Randone S, Martinović D, et al. ”To be or not to be”, ten years after: evidence for mixed connective tissue disease as a distinct entity. Semin Arthritis Rheum. 2012;41(4):589–598. doi:10.1016/j.semarthrit.2011.07.010
- Ciancio N, Pavone M, Torrisi SE, et al. Contribution of pulmonary function tests (PFTs) to the diagnosis and follow up of connective tissue diseases. Multidiscip Respir Med. 2019;15:14–17.
- Maddison PJ, Reichlin M. Quantitation of precipitating antibodies to certain soluble nuclear antigens in SLE. Arthritis Rheum. 1977;20(3):819–824. doi:10.1002/art.1780200310
- Chan EKL, von Muhlen CA, Fritzler MJ, et al. The international Consensus on ANA patterns (ICAP) in 2021- the 6th workshop and current perspectives. J Appl Lab Med. 2022;7(1):322–330. doi:10.1093/jalm/jfab140
- Arbuckle MR, McClain MT, Rubertone MV, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Eng J Med. 2003;349(16):1526–1533. doi:10.1056/NEJMoa021933
- Usuba FS, Lopez JB, Guller R, et al. Sjӧgren’s syndrome: an underdiagnosed condition in mixed connective tissue disease. Clinics. 2014;69(3):158–162. doi:10.6061/clinics/2014(03)02
- Pizzorni C, Ferrari G, Schenone C, et al. Capillaroscopic analysis of the microvascular status in mixed versus undifferentiated connective tissue disease. Microvasc Res. 2022;142:104367. doi:10.1016/j.mvr.2022.104367
- De Holanda Mafaldo Diofenes A, Bonfà E, Fuller R, Correia Caleiro MT. Capillaroscopy is a dynamic process in mixed connective tissue disease. Lupus. 2007;16(4):254–258. doi:10.1177/0961203307076517
- Todoroki Y, Kubo S, Nakano K, et al. Nailfold microvascular abnormalities are associated with a higher prevalence of pulmonary arterial hypertension in patients with MCTD. Rheumatology. 2022;61(12):4875–4884. doi:10.1093/rheumatology/keac165
- Sambataro D, Sambataro G, Libra A, et al. Nailfold videocapillaroscopy is a useful tool to recognize definite forms of systemic sclerosis and idiopathic inflammatory myositis in interstitial lung disease patients. Diagnostics. 2020;10(5):253. doi:10.3390/diagnostics10050253
- Manfredi A, Sebastiani M, Cassone G, et al. Nailfold Capillaroscopic changes in dermatomyositis and polymyositis. Clin Rheumatol. 2015;34(2):279–284. doi:10.1007/s10067-014-2795-8
- Kawano-Dourado L, Baldi BG, Kay FU, et al. Pulmonary involvement in long-term mixed connective tissue disease: functional trends and image findings after 10 years. Clin Exp Rheumatol. 2015;33(2):234–240.
- Wijsenbeek M, Cottin V. Spectrum of fibrotic lung disease. N Eng J Med. 2020;383(10):958. doi:10.1056/NEJMra2005230
- Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Eng J Med. 2019;381(19):1718. doi:10.1056/NEJMoa1908681
- Sambataro G, Vancheri A, Torrisi SE, et al. The morphological domain does not affect the rate of progression to defined autoimmune diseases in patients with interstitial pneumonia with autoimmune features. Chest. 2020;157(1):238–242. doi:10.1016/j.chest.2019.08.2175
- Sambataro G, Ferrara CA, Torrisi SE, et al. “Usual” Interstitial pneumonia with autoimmune features: a prospective study on a cohort of idiopathic pulmonary fibrosis patients. Clin Exp Rheumatol. 2022;40(7):1324–1329. doi:10.55563/clinexprheumatol/lqi6z7
- Sambataro G, Sambataro D, Spicuzza L, et al. Progression and prognosis of interstitial pneumonia with autoimmune features: a longitudinal, prospective, multi-centre study. Clin Exp Rheumatol. 2022. doi:10.55563/clinexprheumatol/lycdca
- Palmucci S, Galioto F, Fazio G, et al. Clinical and radiological features of lung disorders related to connective-tissue diseases: a pictorial assay. Insights Imaging. 2022;13:108. doi:10.1186/s13244-022-01243-2
- Topyla-çPutowska W, Tomaszewski M, Wysokinski A, Tomaszewski A. Echocardiography in pulmonary arterial hypertension: comprehensive evaluation and technical considerations. J Clin Med. 2021;10(15):3229. doi:10.3390/jcm10153229
- Bournia VK, Tsangaris I, Rallidis L, et al. Cardiac catheterization versus echocardiography for monitoring pulmonary pressure: a prospective study in patients with connective tissue disease-associated pulmonary artery hypertension. Diagnostics. 2020;10(1):49. doi:10.3390/diagnostics10010049