
Abstract
Globoid cell leukodystrophy or Krabbe is a disease that affects children as well as adults who have mutations in the gene encoding the enzyme galactosylceramidase/galctocerebrosidase (GALC), resulting in the deposition of the toxic lipid D-galactosyl-beta1-1’ sphingosine (GalSph or psychosine). Several therapeutic modalities were used to treat patients with Krabbe disease, including hematopoietic stem cell transplantation, enzyme replacement therapy, autophagy activators, intravenous immunoglobulin, and inhibitors of the Pyroptosis process, among many other approaches. In this article, I will briefly discuss the disease in both human and animal model, describe recent clinical observations as well as methods utilizing genetic analysis for diagnosis, and finally review recent advances in treating this rare and devastating disease.
Introduction
About 20 years ago, we reported that D-galactosyl-beta1-1’ sphingosine (GalSph, also known as psychosine), and D-glucosyl-beta1-1’ sphingosine (GluSph) induced the in vitro apoptosis of human natural killer (NK) cells.Citation1 NK cells are part of innate lymphoid cells 1 (ILC 1), which play major roles in fighting cancer but are also involved in inflammation.Citation2 Globoid cell leukodystrophy (GLD) or Krabbe disease (KD), is an autosomal recessive disease that affects infants.Citation3–5 The disease is caused by the destruction of oligodendrocytes, reduced myelin formation, and leading to the accumulation of globoid cells.Citation6,Citation7 The toxic lipid GalSph, which is not present in normal brain or other tissues,Citation8 accumulates at high concentrations in the brain of Krabbe patients, due to the deficiency of the enzyme galactosylceramidase/galctocerebrosidase (GALC). Although children are affected by the disease, adult-onset GLD is also prevalent but is usually neglected by clinicians.Citation9 GALC physiology, GLD pathophysiology, and therapeutic strategies for KD were recently reviewed.Citation10
Demyelination was observed in infantile KD patients suffering from GALC deficiency. In addition, an increase of pathological CD8+ T lymphocytes was observed, suggesting a linkage between clinical severity and neuroimmunology. Taken together, these findings support the interplay among proteins in KD brain white matter.Citation11 Microscopic analyses show profound neuro-axonal degeneration with a mild effect on myelin structure, indicating that GALC accumulating in the neurons is essential to protect the function of neurons regardless of myelin, which contributes to the pathogenesis of KD.Citation12 However, it must be noted that although the pathology of the infantile type of Krabbe disease exhibits atrophy of the brain and extensive loss of myelin in the white matter with massive gliosis and multiple globoid cells, in the few cases reporting adult-onset Krabbe disease, a focal and mild demyelination and a few globoid-like cells were observed.
The Disease
Krabbe Disease in Humans
Diagnosis of patients through newborn screening is essential for better quality of life.Citation13 During earlier years, the diagnosis was done by measuring GALC activity in leukocytes and cultured cells. Afterward, newborn screening (NBS) was used. One parameter for the disease diagnosis is the low GALC activity.Citation14 The KD NBS council suggested recommendations for successful classification of NBS positive patients with a prediction of those at risk to progress towards late onset disease.Citation15 As predicted, these programs utilize GALC as an initial test, with an increase in psychosine levels as a confirmatory marker for predicting the onset of KD. Increased of psychosine activity results in the death of oligodendrocytes leading to a sequence of events in the nervous system that lead to the appearance of clinical symptoms associated with the disease.Citation16 However, these testes should be carefully evaluated as there might be a genuine risk of a false positive rate.Citation17
Lactosylceramide metabolism is impaired in fibroblasts isolated from patients with KD in the absence of psychosine, suggesting that molecules other than psychosine might also be involved in the disease.Citation18 In a related matter, it was demonstrated that α-synuclein which is involved in Parkinson’s disease, might also have pathological similarities observed in KD. Consequently, KD brain accumulated α-synuclein leading to fibrils aggregation, suggesting that α-synuclein is involved in KD which could be a target for intervention.Citation19
A KD pluripotent stem cells (iPSCs) line known as PUMCi002-A was generated from dermal fibroblasts of a Krabbe patient’s father with a c.461C>A mutation in GALC gene. The pluripotency, in vitro differentiation potential and karyotype stability of this cell line were confirmed. This cell line can be used to better understand the mechanisms involved in GALC-associated KD, and more important it may provide plausible new therapeutic directions.Citation20
Globoid Cell Leukodystrophy (GLD) in Mice
A natural authentic model for human KD, is the twitcher mouse, which have mutations in their GALC gene,Citation21,Citation22 associated with the deregulation of several proteins.Citation23 GalSph is increased in the kidneys, liver, spleen and highly accumulates in the brains of these animals. A heptahelical receptor that binds heterotrimeric G proteins, named T-cell-death-associated gene 8 (TDAG8), has been shown to bind GalSph and GlcSph.Citation24,Citation25 Human TDAG8 exists in normal tissues and is restricted to lymphoid organs, such as spleen and lymph nodes, as well as its expression in peripheral blood lymphocytes. We previously demonstrated that NK cells express TDAG8 and reported that GalSph and GlcSph damage these cells after binding this receptor.Citation1 In addition, GalSph inhibited NK cell distribution into the spleens of twitcher mice, along with inducing their apoptosis.Citation26 In vivo electrophysiological recordings showed defective basic functional properties of twitcher mice primary visual cortex.Citation27
One protocol describing the genotype determination of GALC mutation status in twitcher mice was based on the allele-discrimination RT-PCR reaction. This method diagnosed GLD accurately, and without ambiguity in homozygotes, wild type, and heterozygotes animals.Citation28 Another study investigated the mechanotransduction process in primary fibroblasts collected from twitcher mice. This study reported that focal adhesions, the protein clusters necessary to adhere and migrate were increased, and accordingly, cell migration and wound healing were affected in these mice.Citation29
Genetic Studies of Krabbe Disease
Heterozygous missense mutation: c.1658G>A (p.G553E) and c.1901T>C (p.L634S) were identified in the GALC gene by whole-exome sequencing. In this study, a certain novel mutation, namely c.1658G>A (p.G553E) was observed, which broadened the mutation spectrum in KD patients.Citation30 Using CRISPR-Cas9 gene editing, point mutations T513M observed in infants introduced in the murine GALC, resulting in GALC T513M/T513M mice. These animals suffer from a decrease in GALC activity, increased psychosine levels, leading to progress towards lethal neurological phenotype. In contrast, another variant, ie, GALC G41S/G41S mice have normal lifespan, modest decreases of GALC, and minimal psychosine accumulation.Citation22
Changes in the gene expression of iPSCs and iPSC-derived neural stem cells (NSCs) from a patient with GLD designated as K-iPSCs/NSCs and normal control designated as AF-iPSCs/NSCs were investigated in order to determine the potential mechanism underlying GLD pathogenesis.Citation31 It was observed that mutations in the GALC gene affected the signaling pathways during neural development, suggesting that alterations in signaling pathways may contribute to GLD pathogenesis. These results also demonstrate that the model based on K-iPSCs may represent a novel tool that can be used to study the underlying molecular basis of GLD.Citation31
Further, astrocytes collected from KD patients contain high levels of glucosylceramide and ceramide, suggesting that in certain cases the disease may not be always associated with the galactosylceramide and psychosine pathways.Citation32 Other neuroinflammatory genes were explored to understand their correlation with disease severity in the twitcher mouse. It was observed that kinases such as Ripk1 kinase does not play a role in KD.Citation33 Collectively, these data suggest that although measuring the levels of GALC and psychosine is essential, other parameters must also be considered in order to reach a conclusive diagnosis therapy before is initiated.
Clinical Observations
It is imperative for pediatricians to identify patients with the disease as soon as possible. However, this might be difficult due to misunderstanding of the disease symptoms.Citation34 Doctors may rely on genetic studies to diagnose the disease. For example, in patients who had a leukodystrophy associated with hypomyelination or delayed myelination on MRI, it was suggested that exome genome sequencing would be used to identify KD patients.Citation35 Further, KD patients with mitochondrial gene NDUFAF1 mutations were observed, suggesting that gene panel examination must be expanded in order not to miss pathogenic mutations.Citation36
In one study, two siblings with atypical clinical and neuroimaging phenotypes of KD were examined, and it was reported that they carry biallelic loss-of-function GALC variants, including a recurrent 30 kb deletion.Citation37 In an infant boy with KD, the pathogenic variants of the disease were screened by whole-exome sequencing. These variants expressed different outcomes. For example, c.328+5G>T variant was predicted to alter splicing, whereas the variant c.658C>T indicates truncation of the protein.Citation38 A whole-exome sequencing identified a pathogenic homozygous missense mutation of the GALC gene in a young woman with an onset KD who was presented with generalized seizures, progressive cognitive decline, psychiatric symptoms, gait ataxia, and action-induced myoclonus.Citation39 Another polymorphic variant known as p.Ile562Thr over-represented in the KD population, was associated with reduced mature GALC protein and activity.Citation11
A patient who developed KD in late infancy had a pathogenic genotype, showed reduced enzyme activity but surprisingly low psychosine levels, suggesting that measuring psychosine levels should be combined with other measurements, such as enzyme levels, as well as genotyping and correlation with the patient’s clinical presentations.Citation40 Therefore, magnetic resonance (MR) images showing abnormal signals, as well as the presence of globoid cells must be done before final diagnosis of the disease.Citation41 For instance, MR imaging diagnosed A 32-year-old woman with headache as adult onset KB with two pathogenic variants in GALC.Citation42
There was a high increase in psychosine levels in infants diagnosed with KD. This was corroborated with increased levels of acid ceramidase, a key enzyme for psychosine production, and hyperglycosylated lysosomal-associated membrane protein 1, a marker for lysosomal activation in periventricular white matter.Citation11 This patient suffered from neuropathological changes. It should be stressed that diagnosis of KB is highly important as the disease carries a tremendous financial burden. A study estimated that KD patients had over $51 million in health care charges and hundreds of hospitalizations, demonstrating the health impacts of KD on the society.Citation43
Role of GALC in Cancer
Lysophospholipids play major roles in tumor microenvironment.Citation44 In this regard, GALC also plays a role in cancer.Citation45 Removal of β-galactose from β-galactosylceramide, leads to the formation of the oncosuppressor metabolite ceramide,Citation46 which may play a role in tumor growth and differentiation.Citation47 GalSph and GlcSph, lipids that disrupt the lysosomes, play definitive roles in cancer therapy.Citation48 Consequently, they may contribute to the development of multidrug-resistance by cancer cells. It was shown that GlcCer may provoke immune reaction and acts as a self-antigen in KD. On the other hand, GalCer was recognized as an important cellular receptor for HIV-1.Citation49
Therapeutic Approaches for Treatment of Krabbe Disease
Early studies showed that transplantation of hematopoietic stem cells from normal mice into syngeneic twitcher mice resulted in increased survival, but in these studies the success of the transplantation procedures did not go beyond 100 days.Citation50,Citation51 In five patients with late-onset GLD, a high successful rate of recovery after hematopoietic stem cell transplantation was reported.Citation52 A comprehensive study examining the impact of hematopoietic stem cell transplantation (HSCT), demonstrated that this therapeutic regimen is associated with reduced mortality in KD patients.Citation53 In a case study of KD patient showing progressive spastic paraparesis who underwent HSCT, it was observed that GALC activity returned to a normal level and the lesions in the brain and spinal cord became faint on images.Citation54 Among patients between 24 and 40 days of age, HSCT therapy was successful and the patients were alive 30 to 58 months later.Citation13 However, HSCT must be done during the first weeks of life before symptoms develop.
Another regimen of therapy utilized recombinant adeno-associated virus (rAAV) vectors to infect Krabbe patient-derived neural stem cells (K-NSCs). Using several infected K-NSCs, this therapeutic regimen rescued GALC enzymatic activity in infected cells.Citation55 Another study successfully showed that human KD patients can be treated with high-dose AAVrh10 without blood stem cell transplantation.Citation56 The efficacy and safety of a single cisterna magna AAVhu68 administration to treat KD was also reported.Citation57 In twitcher mice, adenovirus gene therapy of the lysosomal enzyme GALC significantly ameliorated central and peripheral neuropathology, prolonged survival, and largely normalized motor deficits. These observations indicate that AAV-gene therapy may be considered an alternative mode of effective intervention.Citation58
Rapamycin, an inhibitor of mTOR but autophagy activator, was used in twitcher mice in order to reduce the deposition of insoluble ubiquitinated protein, corroborated with the attenuation of astrogliosis and microgliosis.Citation59 This therapeutic modality was critical in inducing cortical myelination, neurite density, and rescued the neurological abnormalities of twitcher mice. Further, it induced cell migration and improved the clearance of focal adhesion in GALC-deficient fibroblasts.Citation60 Another therapeutic approach utilized lecithin/chitosan nanoparticles to prevent the cytotoxicity caused by psychosine in cultured human astrocytes in vitro suggesting a direct interaction between psychosine and the nanoparticles.Citation61
Enzyme replacement therapy is one of the initial approaches used to treat KD patients. However, it exerted limitations in relieving CNS lesions due to difficulties in passing the blood–brain barrier.Citation62 Administration of the enzyme GALC inserted into adeno-virus to twitcher mice resulted in the accumulation of this enzyme in the brains of these mice which consequently, led to prolong the mice lives.Citation63 Treating twitcher mouse with S202 amide, a galactosyltransferase inhibitor reduced GalCer and psychosine concentrations in the central and peripheral nervous systems, which significantly increased lifespan, although further studies are needed to understand the full therapeutic potential of such an inhibitor.Citation64 Substrate reduction therapy was also investigated utilizing RA 5557, a brain-penetrant inhibitor of GALC biosynthesis, in twitcher mice that lack GALC activity. This modality was effective and has an acceptable safety in vivo profile.Citation65
Intriguingly, a novel approach for therapy was developed. Here, a young girl who was diagnosed with KD, received intravenous immunoglobulin (IVIg), and upon which her limb weakness improved. This was repeated when she reached 16 years old, and again her symptoms gradually improved. It was suggested that in certain KB cases, IVIg may be considered as a form of therapy.Citation66
Pentraxin-3 (PTX3), a soluble pattern recognition receptor and a regulator of innate immunity,Citation67 was seen in the CNS of GALC-deficient Krabbe patients and twitcher mice. PTX3 may exert a protective role by reducing the neuroinflammatory response that occurs in the spinal cord of GALC-deficient animals. Regarding the role of innate immune system, in situ hybridization and immunohistochemical staining revealed that the expression of pro-inflammatory non-canonical caspase-11, canonical caspase-1, gasdermin D and cognate genes are induced in the nervous tissues. Caspase-11 was also found in reactive microglia/macrophages as well as in astrocytes, but caspase-1 and gasdermin D were restricted to reactive microglia/macrophages.Citation68 These results suggest that innate immune system might be involved in the inflammatory reactions associated with KB and twitcher mice. The role of the Pyroptosis process which utilizes gasdermin D and inflammasomes, in linking innate and adaptive immunity has been described.Citation69,Citation70 It is therefore suggested that inhibitors of Pyroptosis such as those that inhibit inflammasomes, caspases, gasdermins, among others,Citation71,Citation72 should be evaluated for effective intervention in Krabbe disease.
Concluding Remarks
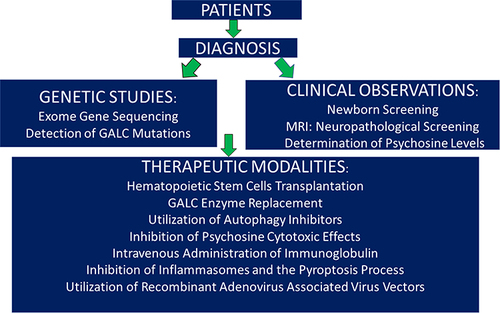
Although Krabbe disease was discovered by Dr Krabbe in the first twenty years of the last century, advances in understanding and treating this disease have been slow. When patients are presented in the clinic with a suspicious of disease occurring, neurologists and physicians rely on genetic studies in addition to the clinical observations in order to confirm the disease occurrence. Once this is validated, treatment can be started. Albeit early therapeutic modalities utilized hematopoietic stem cells transplantation and GALC enzyme replacement which are costly, recently several new therapeutic regimens have been successfully used to treat the disease, as shown in . This figure also describes the algorithm of human disease from the time the patients enter the clinic until the time of therapy. The choice of the therapeutic modality should be agreed upon between the physicians and the parents of the patients diagnosed with the disease. Finally, the discovery of twitcher mice which resemble globoid cell leukodystrophy in humans, aided to some extent in certain aspects, particularly in generating novel therapeutic modalities. In this short review, I briefly touched on the nature of the disease and discussed novel approaches using genetic methods. I also discussed recent advances in treating KD.
Figure 1 Algorithm of human Krabbe disease.
Disclosure
The author reports no conflicts of interest in this work.
References
- Maghazachi AA, Knudsen E, Jin Y, Jenstad M, Chaudhry FA. D-galactosyl-beta1-1’-sphingosine and D-glucosyl-beta1-1’-sphingosine induce human natural killer cell apoptosis. Biochem Biophys Res Commun. 2004;320(3):810–815. doi:10.1016/j.bbrc.2004.06.027
- Elemam NM, Hannawi S, Maghazachi AA. Innate lymphoid cells (ILCs) as mediators of inflammation, release of cytokines and lytic molecules. Toxins. 2017;9(12):398. doi:10.3390/toxins9120398
- Krabbe K. A new familial, infantile form of diffuse brain sclerosis. Brain. 1916;30:74–114. doi:10.1093/brain/39.1-2.74
- Svennerholm L, Vanier M-T, Månsson JE. Krabbe disease: a galactosylsphingosine (psychosine) lipidosis. J Lipid Res. 1980;21(1):53–64. doi:10.1016/S0022-2275(20)39839-4
- Vanier M-T, Svennerholm L. Chemical pathology of Krabbe’s disease. III. Ceramide hexosides and gangliosides of brain. Acta Paediatr Scand. 1975;64(4):641–648. doi:10.1111/j.1651-2227.1975.tb03896.x
- Baskin GB, Ratterree M, Davison BB, et al. Genetic galactocerebrosidase deficiency (globoid cell leukodystrophy, Krabbe disease) in rhesus monkeys (Macaca mulatta). Lab Anim Sci. 1998;48(5):476–482.
- Wenger DA. Murine, canine and non-human primate models of Krabbe disease. Mol Med Today. 2000;6(11):449–451. doi:10.1016/S1357-4310(00)01800-1
- Duchen LW, Eicher EM, Jacobs JM, Scaravilli F, Teixeira F. Hereditary leucodystrophy in the mouse: the new mutant twitcher. Brain. 1980;103(3):695–710. doi:10.1093/brain/103.3.695
- Wu G, Li Z, Li J, et al. A neglected neurodegenerative disease: adult-onset globoid cell leukodystrophy. Front Neurosci. 2022;16:998275. doi:10.3389/fnins.2022.998275
- Feltri ML, Weinstock NI, Favret J, Dhimal N, Wrabetz L, Shin D. Mechanisms of demyelination and neurodegeneration in globoid cell leukodystrophy. Glia. 2021;69(10):2309–2331. doi:10.1002/glia.24008
- Iacono D, Koga S, Peng H, et al. Galactosylceramidase deficiency and pathological abnormalities in cerebral white matter of Krabbe disease. Neurobiol Dis. 2022;174:105862. doi:10.1016/j.nbd.2022.105862
- Kreher C, Favret J, Weinstock NI, et al. Neuron-specific ablation of the Krabbe disease gene galactosylceramidase in mice results in neurodegeneration. PLoS Biol. 2022;20(7):e3001661. doi:10.1371/journal.pbio.3001661
- Page KM, Ream MA, Rangarajan HG, et al. Benefits of newborn screening and hematopoietic cell transplant in infantile Krabbe disease. Blood Adv. 2022;6(9):2947–2956. doi:10.1182/bloodadvances.2021006094
- Wenger DA, Luzi P, Rafi MA. Advances in the diagnosis and treatment of Krabbe disease. Int J Neonatal Screen. 2021;7(3):57. doi:10.3390/ijns7030057
- Thompson-Stone R, Ream MA, Gelb M, et al. Consensus recommendations for the classification and long-term follow up of infants who screen positive for Krabbe Disease. Mol Genet Metab. 2021;134(1–2):53–59. doi:10.1016/j.ymgme.2021.03.016
- Ezer S, Zuckerman S, Segel R, Zlotogora J. Carrier screening for Krabbe disease in an isolated inbred community. Am J Med Genet. 2022;188(9):2555–2559. doi:10.1002/ajmg.a.62882
- Jalal K, Carter RL, Barczykowski A, Tomatsu S, Langan TJ. A Roadmap for potential improvement of newborn screening for inherited metabolic diseases following recent developments and successful applications of bivariate normal limits for pre-symptomatic detection of MPS I, Pompe Disease, and Krabbe disease. Int J Neonatal Screen. 2022;8(4):61. doi:10.3390/ijns8040061
- Papini N, Giallanza C, Brioschi L, et al. Galactocerebrosidase deficiency induces an increase in lactosylceramide content: a new hallmark of Krabbe disease? Int J Biochem Cell Biol. 2022;145:106184. doi:10.1016/j.biocel.2022.106184
- Hatton C, Ghanem SS, Koss DJ, Abdi IY, Gibbons E, Guerreiro R. Prion-like α-synuclein pathology in the brain of infants with Krabbe disease. Brain. 2022;145(4):1257–1263. doi:10.1093/brain/awac002
- Lv YF, Wang J, Cao CY, Zhang Y, Wang W. Production and characterization of human induced pluripotent stem cell line (PUMCi002-A) from a Krabbe patient related control to study disease mechanisms associated with GALC mutation. Stem Cell Res. 2022;65:102945. doi:10.1016/j.scr.2022.102945
- Tanaka K, Nagara H, Kobayashi T, Goto I. The twitcher mouse: accumulation of galactosylsphingosine and pathology of sciatic nerve. Brain Res. 1988;454(1–2):340–346. doi:10.1016/0006-8993(88)90835-9
- Rebiai R, Rue E, Zaldua S, et al. CRISPR-Cas9 Knock-In of T513M and G41S Mutations in the murine β-Galactosyl-Ceramidase gene re-capitulates early-onset and adult-onset forms of Krabbe disease. Front Mol Neurosci. 2022;15:896314. doi:10.3389/fnmol.2022.896314
- Vantaggiato L, Shaba E, Carleo A, et al. Neurodegenerative disorder risk in Krabbe disease carriers. Int J Mol Sci. 2022;23(21):13537. doi:10.3390/ijms232113537
- Choi JW, Lee SY, Choi Y. Identification of a putative G protein-coupled receptor induced during activation-induced apoptosis of T cells. Cell Immunol. 1996;168(1):78–84. doi:10.1006/cimm.1996.0051
- Kyaw H, Zeng Z, Su K, et al. Cloning, characterization, and mapping of human homolog of mouse T-cell death-associated gene. DNA Cell Biol. 1998;17(6):493–500. doi:10.1089/dna.1998.17.493
- Al-Falahi Y, Sand KL, Knudsen E, Damaj BB, Rolin J, Maghazachi AA. Splenic natural killer cell activity in two models of experimental neurodegenerative diseases. J Cell Mol Med. 2009;13:2693–2703. doi:10.1111/j.1582-4934.2008.00640.x
- Tonazzini I, Cerri C, Del Grosso A, et al. Visual system impairment in a mouse model of Krabbe disease: the twitcher mouse. Biomolecules. 2020;23(1):7. doi:10.3390/biom11010007
- Carpi S, Del Grosso A, De Sarlo M, et al. Reliable and fast genotyping protocol for Galactosylceramidase (Galc) in the twitcher (Twi) mouse. Biomedicines. 2022;10(12):3146. doi:10.3390/biomedicines10123146
- Mezzena R, Del Grosso A, Pellegrino RM, et al. Mechanotransduction impairment in primary fibroblast model of Krabbe disease. Biomedicines. 2023;11(3):927. doi:10.3390/biomedicines11030927
- He Z, Pang X, Bai J, et al. A novel GALC gene mutation associated with adult-onset Krabbe disease: a case report. Neurocase. 2022;28(3):314–319. doi:10.1080/13554794.2022.2083518
- Lv Y, Qin Y, Wang J, et al. Identifying altered developmental pathways in human globoid cell leukodystrophy iPSCs-derived NSCs using transcriptome profiling. BMC Genomics. 2023;24(1):210. doi:10.1186/s12864-023-09285-6
- Lieberman R, Cortes LK, Gao G, et al. Human iPSC-derived astrocytes generated from donors with globoid cell leukodystrophy display phenotypes associated with disease. PLoS One. 2022;17(8):e0271360. doi:10.1371/journal.pone.0271360
- Cachón-González MB, Wang S, Cox TM. Expression of Ripk1 and DAM genes correlates with severity and progression of Krabbe disease. Hum Mol Genet. 2021;30(22):2082–2099. doi:10.1093/hmg/ddab159
- Modesti NB, Evans SH, Jaffe N, Vanderver A, Gavazzi F. Early recognition of patients with leukodystrophies. Current Problems in Pediatric and Adolescent Health Care. 2022;52(12):101311. doi:10.1016/j.cppeds.2022.101311
- Perrier S, Guerrero K, Tran LT, et al. Solving inherited white matter disorder etiologies in the neurology clinic: challenges and lessons learned using next-generation sequencing. Front Neurol. 2023;14:1148377. doi:10.3389/fneur.2023.1148377
- Wu L, Liao X, Yang S, Gan S. Krabbe disease associated with mitochondrial dysfunction in a Chinese family. Front Neurol. 2021;12:750095. doi:10.3389/fneur.2021.750095
- Nicita F, Stregapede F, Deodato F, et al. “Atypical” Krabbe disease in two siblings harboring biallelic GALC mutations including a deep intronic variant. Eur J Hum Genet. 2022;30(8):984–988. doi:10.1038/s41431-022-01111-z
- Zhang X, Niu G, Song P, et al. Compound heterozygous pathogenic variants in the GALC gene cause infant-onset Krabbe disease. Transl Pediatr. 2021;10(10):2552–2562. doi:10.21037/tp-21-403
- Wang Y, Wang SY, Li K, et al. Adult-onset Krabbe disease presenting with progressive myoclonic epilepsy and asymmetric occipital lesions: a case report. Front Neurol. 2022;13:1010150. doi:10.3389/fneur.2022.1010150
- Corre CS, Matern D, Pellegrino JE, Saavedra-Matiz CA, Orsini JJ, Thompson-Stone R. Low psychosine in Krabbe disease with onset in late infancy: a case report. Int J Neonatal Screen. 2021;7(2):28. doi:10.3390/ijns7020028
- Jaiswani AK, Kulkarni V, Paliwal A. Krabbe’s disease; A rare case report. Leg Med. 2023;60:102155. doi:10.1016/j.legalmed.2022.102155
- Paiva ARB, Fonseca Neto RE, Afonso CL, Freua F, Nóbrega PR, Kok F. Incidental magnetic resonance imaging findings leading to an unusual diagnosis: adult onset Krabbe disease. Eur J Neurol. 2022;29(6):1859–1862. doi:10.1111/ene.15298
- Ghabash G, Wilkes J, Barney BJ, Bonkowsky JL. Hospitalization burden and incidence of Krabbe disease. J Child Neurol. 2022;37(1):12–19. doi:10.1177/08830738211027717
- Rolin J, Maghazachi AA. Effects of lysophospholipids on tumor microenvironment. Cancer Microenviron. 2011;4(3):393–403. doi:10.1007/s12307-011-0088-1
- Belleri M, Presta M. β-Galactosylceramidase in cancer: more than a psychosine scavenger. Oncoscience. 2022;9:11–12. doi:10.18632/oncoscience
- Belleri M, Chiodelli P, Corli M, Capra M, Presta M. Oncosuppressive and oncogenic activity of the sphingolipid-metabolizing enzyme β-galactosylceramidase. Biochim Biophys Acta Rev Cancer. 2022;1877(1):188675. doi:10.1016/j.bbcan.2021
- Reiter CR, Rebiai R, Kwak A, et al. The pathogenic sphingolipid psychosine is secreted in extracellular vesicles in the brain of a mouse model of Krabbe disease. ASN Neuro. 2022;14:17590914221087817. doi:10.1177/17590914221087817
- Stahl-Meyer K, Bilgin M, Holland LKK, et al. Galactosyl- and glucosylsphingosine induce lysosomal membrane permeabilization and cell death in cancer cells. PLoS One. 2022;17(11):e0277058. doi:10.1371/journal.pone.0277058
- Abed Rabbo M, Khodour Y, Kaguni LS, Stiban J. Sphingolipid lysosomal storage diseases: from bench to bedside. Lipids Health Dis. 2021;20(1):44. doi:10.1186/s12944-021-01466-0
- Ichioka T, Kishimoto Y, Brennan S, Santos GW, Yeager AM. Hematopoietic cell transplantation in murine globoid cell leukodystrophy (the twitcher mouse): effects on levels of galactosylceramidase, psychosine, and galactocerebrosides. Proc Natl Acad Sci USA. 1987;84(12):4259–4263. doi:10.1073/pnas.84.12.4259
- Yeager AM, Brennan S, Tiffany C, Moser HW, Santos GW. Prolonged survival and remyelination after hematopoietic cell transplantation in the twitcher mouse. Science. 1984;225(4666):1052–1054. doi:10.1126/science.6382609
- Krivit W, Shapiro EG, Peters C, et al. Hematopoietic stem-cell transplantation in globoid-cell leukodystrophy. N Engl J Med. 1998;338(16):1119–1126. doi:10.1056/NEJM199804163381605
- Ghabash G, Wilkes J, Bonkowsky JL. National U.S. patient and transplant data for Krabbe disease. Front Pediatr. 2021;9:764626. doi:10.3389/fped.2021.76462
- Mitsutake A, Matsukawa T, Iwata A, et al. Favorable outcome of hematopoietic stem cell transplantation in late-onset Krabbe disease. Brain Dev. 2023;S0:387–7604(23)00066–9. doi:10.1016/j.braindev.2023.04.001
- Tian G, Cao C, Li S, Wang W, Zhang Y, Lv Y. rAAV2-mediated restoration of GALC in neural stem cells from Krabbe patient-derived iPSCs. Pharmaceuticals. 2023;16(4):624. doi:10.3390/ph1604062
- Rafi MA, Luzi P, Wenger DA. Can early treatment of twitcher mice with high dose AAVrh10-GALC eliminate the need for BMT? Bioimpacts. 2021;11(2):135–146. doi:10.34172/bi.2021.21
- Hordeaux J, Jeffrey BA, Jian J, et al. Efficacy and safety of a Krabbe disease gene therapy. Hum Gene Ther. 2022;33(9–10):499–517. doi:10.1089/hum.2021.245
- Heller GJ, Marshall MS, Issa Y, et al. Waning efficacy in a long-term AAV-mediated gene therapy study in the murine model of Krabbe disease. Mol Ther. 2021;29(5):1883–1902. doi:10.1016/j.ymthe.2021.01.026
- Lin DS, Huang YW, Lee TH, et al. Rapamycin alleviates protein aggregates, reduces neuroinflammation, and rescues demyelination in globoid Cell leukodystrophy. Cells. 2023;12(7):993. doi:10.3390/cells12070993
- Mezzena R, Del Grosso A, Pellegrino RM, et al. Mechanotransduction Impairment in primary fibroblast model of Krabbe disease. Biomedicines. 2023;11(3):927. doi:10.3390/biomedicines11030927
- Clementino A, Velasco-Estevez M, Buttini F, Sonvico F, Dev KK. Hybrid nanoparticles as a novel tool for regulating psychosine-induced neuroinflammation and demyelination In vitro and ex vivo. Neurotherapeutics. 2021;18(4):2608–2622. doi:10.1007/s13311-021-01109-3
- Zhou H, Wu Z, Wang Y, et al. Rare diseases in glycosphingolipid metabolism. Adv Exp Med Biol. 2022;1372:189–213. doi:10.1007/978-981-19-0394-6_13
- Rafi MA, Rao HZ, Luzi P, Curtis MT, Wenger DA. Extended normal life after AAVrh10-mediated gene therapy in the mouse model of Krabbe disease. Mol Ther. 2012;20(11):2031–2042. doi:10.1038/mt.2012.153
- Babcock MC, Mikulka CR, Wang B, et al. Substrate reduction therapy for Krabbe disease and metachromatic leukodystrophy using a novel ceramide galactosyltransferase inhibitor. Sci Rep. 2021;11(1):14486. doi:10.1038/s41598-021-93601-1
- Zaccariotto E, Cachón-González MB, Wang B, et al. A novel brain-penetrant oral UGT8 inhibitor decreases in vivo galactosphingolipid biosynthesis in murine Krabbe disease. Biomed Pharmacother. 2022;149:112808. doi:10.1016/j.biopha.2022.112808
- Fukazawa R, Takeuchi H, Oka N, Shibuya T, Sakai N, Fujii A. Adult Krabbe disease that was successfully treated with intravenous immunoglobulin. Intern Med. 2021;60(8):1283–1286. doi:10.2169/internalmedicine.6094-20
- Coltrini D, Chandran AMK, Belleri M, et al. β-Galactosylceramidase deficiency causes upregulation of long Pentraxin-3 in the central nervous system of Krabbe patients and twitcher mice. Int J Mol Sci. 2022;23(16):9436. doi:10.3390/ijms23169436
- Cachón-González MB, Zhao C, Franklin RJ, Cox TM. Upregulation of non-canonical and canonical inflammasome genes associates with pathological features in Krabbe disease and related disorders. Hum Mol Genet. 2023;32(8):1361–1379. doi:10.1093/hmg/ddac299
- Muhammad JS, Jayakumar MN, Elemam NM, et al. Gasdermin D Hypermethylation Inhibits Pyroptosis And LPS-Induced IL-1β Release From NK92 Cells. Immunotargets Ther. 2019;8:29–41. doi:10.2147/ITT.S219867
- Hachim MY, Khalil BA, Elemam NM, Maghazachi AA. Pyroptosis: the missing puzzle among innate and adaptive immunity crosstalk. J Leukoc Biol. 2020;108(1):323–338. doi:10.1002/JLB.3MIR0120-625R
- Demarco B, Danielli S, Fischer FA, Bezbradica JS. How Pyroptosis contributes to inflammation and fibroblast-macrophage cross-talk in rheumatoid arthritis. Cells. 2022;11(8):1307. doi:10.3390/cells11081307
- Tan Y, Chen Q, Li X, et al. Pyroptosis: a new paradigm of cell death for fighting against cancer. J Exp Clin Cancer Res. 2021;40(1):153. doi:10.1186/s13046-021-01959-x