
Abstract
Alzheimer’s disease (AD) is a fatal neurodegenerative disease with a subtle and progressive onset and is the most common type of dementia. However, its etiology and pathogenesis have not yet been fully elucidated. The common pathological manifestations of AD include extraneuronal β-amyloid deposition (Aβ), intraneuronal tau protein phosphorylation leading to the formation of ‘neurofibrillary tangles’ (NFTs), neuroinflammation, progressive loss of brain neurons/synapses, and glucose metabolism disorders. Current treatment approaches for AD primarily focus on the ‘Aβ cascade hypothesis and abnormal aggregation of hyperphosphorylation of tau proteins’, but have shown limited efficacy. Therefore, there is an ongoing need to identify more effective treatment targets for AD. The central nervous system (CNS) inflammatory response plays a key role in the occurrence and development of AD. Neuroinflammation is an immune response activated by glial cells in the CNS that usually occurs in response to stimuli such as nerve injury, infection and toxins or in response to autoimmunity. Neuroinflammation ranks as the third most prominent pathological feature in AD, following Aβ and NFTs. In recent years, the focus on the role of neuroinflammation and microglia in AD has increased due to the advancements in genome-wide association studies (GWAS) and sequencing technology. Furthermore, research has validated the pivotal role of microglia-mediated neuroinflammation in the progression of AD. Therefore, this article reviews the latest research progress on the role of neuroinflammation triggered by microglia in AD in recent years, aiming to provide a new theoretical basis for further exploring the role of neuroinflammation in the process of AD occurrence and development.
Introduction
Alzheimer’s disease (AD), a progressive neurodegenerative disease, is the most common type of dementia in elderly people. Currently, there are approximately 10 million AD patients in China, and by 2050, this number is expected to increase to more than 40 million. AD is characterized by progressive cognitive decline, mental and behavioral abnormalities, and a reduced ability to perform daily activities.Citation1 The characteristic pathological changes are cortical atrophy and a decrease in the number of memory neurons, accompanied by amyloid-β (Aβ) deposits and neurofibrillary tangles (NFTs).Citation2,Citation3 The etiology and pathogenesis of AD remain unclear, despite an increase in the number of patients with this disease in recent years. The amyloid cascade hypothesis and tau hypothesis are the main explanations of the pathogenic mechanisms of AD.Citation4 However, recent failures in therapeutic strategies targeting Aβ have raised doubts about the accuracy of the Aβ cascade hypothesis. Further research has revealed that neuroinflammation, abnormal calcium regulation, mitochondrial dysfunction, and abnormalities in the autophagic lysosomal degradation pathway are also closely linked to the development of AD.Citation5,Citation6 High levels of neuroinflammatory factors have been observed in the cerebral cortex and cerebrospinal fluid of AD patients, indicating a sustained inflammatory response in their brains. The identification of AD risk genes associated with innate immune function and related pathologies suggests that neuroinflammation plays an important role in the pathogenesis of AD.Citation7,Citation8 High levels of neuroinflammatory factors have been observed in the cerebral cortex and cerebrospinal fluid of AD patients, indicating a sustained inflammatory response in their brainsCitation7. Moreover, hyperactivation of microglia leads to a persistent inflammatory response that contributes to the progression of AD.Citation9
To date, the US Food and Drug Administration has approved a total of seven drugs for treating AD. These include three acetylcholinesterase inhibitors (donepezil, galantamine, and carboplatin), the N-methyl-D-aspartate receptor antagonist memantine hydrochloride, Namzaric, and two anti-Aβ-mab approvals in January 2021 and January 2023—adunizumab and lencanizumab.Citation10 However, it is important to note that these drugs only improve symptoms and do not reverse the condition. Some drugs or therapies have not been validated in clinical trials and may even have a negative impact on cognitive function in AD patients.Citation10 The exact cause of AD is still unknown, and a definitive cure or effective symptom relief has not yet been developed. Neuroinflammation, which plays a key role in AD, could be a promising new target for therapy.Citation11 Microglia are a crucial component of the innate immune system in the central nervous system (CNS), playing a significant role in neuroinflammatory responses. They are involved in various pathological processes related to neurodegenerative diseases and contribute to acute and chronic inflammatory responses, influencing the direction of inflammatory outcomes.Citation12,Citation13 During brain development, microglia and neurons form simultaneously. Microglia support neuron survival and neural circuit formation by releasing neurotrophic factors like insulin-like growth factor-1.Citation14 They also participate in non-inflammatory clearance of damaged neurons through TREM2 signaling-mediated phagocytosis, inducing programmed cell death in neurons.Citation15 Additionally, microglia regulate synaptic density, glutamatergic receptors, and dendritic spines, as well as synaptic plasticity through DAP12 signaling.Citation16,Citation17 Overall, microglia play a crucial role in maintaining CNS homeostasis. Thus, this article reviews recent research on neuroinflammation, particularly focusing on the role of microglia in the development of AD, to stimulate further consideration among researchers regarding potential directions for diagnosing and treating AD.
Microglia and AD
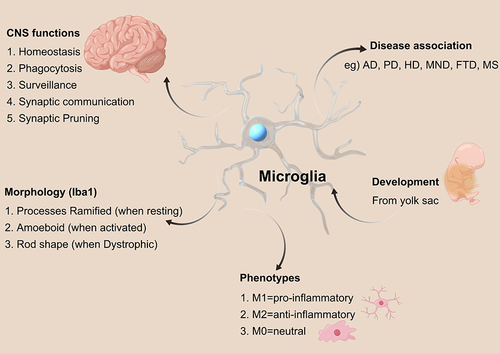
Microglia
Glial cells in the CNS are primarily composed of Microglia, astrocytes, and oligodendrocytes.Citation18 Microglia, which constitute approximately 10% of the total cell population in the CNS, are the predominant mononuclear transitional phagocytes within brain tissue.Citation19,Citation20 The brain is an immune-privileged organ, and microglia are one of the only resident immune cells of the brain, providing the primary immune defense of the CNSCitation21,Citation22 (). Resting state (M0 type) microglia have a branched morphology and play an “immunosurveillance” role. In the activated state, microglia are phagocytic, and when disease or injury occurs, activated microglia can participate in the inflammatory response and immune response as colonized inflammatory cells in the CNS.Citation23,Citation24 Classically activated (M1 type) microglia release proinflammatory factors and toxic substances to kill pathogensCitation20 (). Clinical studies have shown that overactivated M1 microglia can cause neuronal dysfunction, injury, and degeneration and play an important role in AD.Citation25 The alternatively activated (M2) microglia engage in phagocytosis to remove harmful substances such as bacteria, dead cells, and aggregated proteins from the CNS and secrete soluble factors (eg, chemoattractants, cytokines, and neurotrophic factors) to participate in the immune response and repair of damaged brain tissues, thus participating in neuroprotection.Citation19,Citation20
Figure 1 Illustration depicting the associations between neurons and glial cells in the CNS. (The CNS is primarily made up of neurons and glial cells, including astrocytes, oligodendrocytes, and microglia. Glial cells play a crucial role in supporting the survival of neurons by providing them with trophic factors).
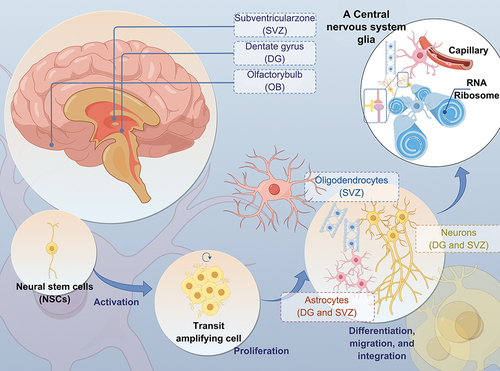
Figure 2 Microglia Function and Communication (Microglia play essential roles in the CNS, such as synaptic pruning, synaptogenesis, axon fasciculation, neurite formation, programmed cell death, astrocyte activation and proliferation, and oligodendrocyte differentiation and myelogenesis).
In recent years, several genome-wide association studies based on clinical AD patient samples have shown that over 50% of confirmed genetic variants in AD-related genes are linked to innate immune function and microglial function. Specifically, genes such as triggering receptor expressed on myeloid cells-2 (TREM2), CD33, apolipoprotein E (APOE) 4, CR1, and HLA-DRB1 have been implicated. Activation of microglial cell surface receptors such as TREM2 and Toll-like receptors in pathological states such as AD has been observed, with these receptors binding to Aβ and APOE and migrating directionally toward sites of injury.Citation26,Citation27 In October 2020, a novel gene, FAM171A2, was discovered to regulate the production of progranulin (PGRN), a precursor of vascular endothelial cells. PGRN, a secreted glycoprotein, plays various roles in neural development, regeneration, neuroinflammation, and autophagy, among other life processes.Citation28,Citation29 Dysregulation of PGRN has been associated with the development of neurodegenerative disorders such as AD, Parkinson’s disease, and frontotemporal lobe dementia.Citation30–33 Furthermore, some studies have proposed that microglia can specifically detect amyloid plaques in the early stages of AD pathogenesis by aggregating around these plaques to create a physical barrier that impedes the spread of neurotoxicity.Citation34 This suggests that microglia-mediated neuroinflammation plays a crucial role in AD pathology and may be linked to the diversity and functional heterogeneity of microglia in the context of ADCitation30 ().
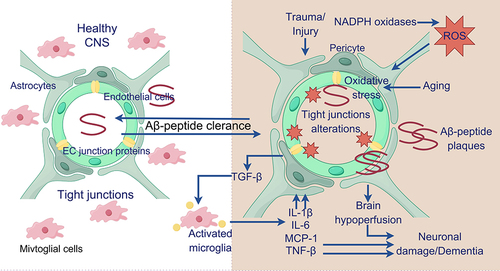
Figure 3 Impact of microglia on AD. (Healthy and young cells, such as microglia, release pro- and anti-inflammatory mediators, growth factors, and bioenergetic pathway mediators. These substances play a role in maintaining brain homeostasis, supporting neuronal survival, and preserving cognitive function).
TREM2 and sTREM2
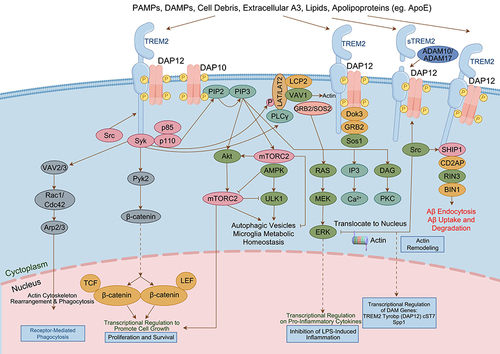
The protein encoded by the TREM2 gene is an intrinsic immune cell receptor expressed on the surface of myeloid cells, and in the brain, it is expressed on microglia;Citation35 this protein is located mainly in the intracellular Golgi apparatus and plays an important role in the regulation of microglial energy metabolism and polarization. TREM2 has now been identified as a potential therapeutic target for AD. Research has shown that a variant of the TREM2, R47H, is strongly associated with AD, and its impact on disease progression is comparable to that of the apolipoprotein E ε4 gene. In vitro, the common variant (CV) of TREM2 has been shown to interact with anionic lipids, while the R47H mutation hinders this binding ability. In this study, researchers created transgenic mice that expressed human CV or R47H TREM2 but lacked endogenous TREM2 in the 5XFAD AD model.Citation36 The results showed that only the CV transgene was able to restore Aβ-induced microgliosis and microglial activation, suggesting that R47H hinders TREM2 function in vivo.Citation36 TREM2 is a single-channel transmembrane receptor belonging to the immunoglobulin superfamily. It consists of an extracellular structural domain with immunoglobulin-containing regions, a transmembrane domain, and a short cytoplasmic tail region.Citation37 This receptor is capable of binding various molecules, such as pathogen-associated molecular patterns (PAMPs), damage-associated molecular patterns (DAMPs), cellular debris, lipids, and apolipoproteins, through its immunoglobulin domains. Activation through cleavage by ADAM family enzymes such as ADAM10 and ADAM17 results in the release of a secreted form of the extracellular domain known as soluble triggering receptor expressed on myeloid cells-2 (sTREM2), halting the signaling cascade associated with TREM2. This cascade is crucial for microglial survival and proinflammatory signaling.Citation38,Citation39 On the other hand, the transmembrane domain of TREM2 can transmit intracellular signals by interacting with a ligand called transmembrane immune signaling adaptor (TYROBP or DAP12). DAP12, a signaling bridging protein expressed in cells involved in the innate immune response. DAP12 contains an immunoreceptor tyrosine-based activation motif that is activated by binding to the TREM2 transmembrane domain, leading to a cascade of intracellular signaling events.Citation40 The association between TREM2 and DAP12 is coordinated by an electrostatic interaction between a lysine in TREM2 and an aspartic acid in DAP12, and once DAP12 binds to TREM2, the tyrosine residue on DAP12 is phosphorylated, causing the recruitment of spleen tyrosine kinase. This triggers a series of tyrosine phosphorylation events that subsequently activate downstream mediators such as phospholipase Cγ2, phosphatidylinositol 3-kinase (PI3K), mammalian target of rapamycin (mTOR) and mitogen-activated protein kinase (MAPK),Citation41,Citation42 leading to cellular activation. Functional studies have demonstrated that the absence of TREM2 hinders proper microglial reactions to damage and cues that typically induce cell movement in various ways. In a brain slice assay conducted ex vivo, the lack of TREM2 diminished the migration distance of microglia.Citation43 In an in vivo experiment, microglia deficient in TREM2 exhibited reduced migration toward injected apoptotic neurons. Additionally, the extension of microglial processes toward areas of laser-induced focal CNS injury in the somatosensory cortex is delayed.Citation43 These findings suggest that alterations in the gene expression of TREM2 and sTREM2 affect microglial function ().
Figure 4 Schematic diagram of TREM2 signaling interactions.
CD33
Research on AD has identified more than 30 genetic loci, many of which are linked to the immune response and microglia.Citation44 Notably, CD33 and TREM2 have been highlighted in these studies.Citation45–49 CD33 encodes a sialic acid-binding immunoglobulin-like lectin found on myeloid progenitor cells, monocytes, and macrophages. It plays a role in cell adhesion and endocytosis, as well as inhibiting cytokine release and regulating immune cell growthCitation50 and TLR4 signaling.Citation51 CD33 is a type I transmembrane protein that acts as an inhibitory immune receptor. It is part of the Siglec family. The external domains of Siglecs can be used to detect glycans containing sialic acid, influencing immune cell signaling pathways via their internal tail.Citation52 CD33 belongs to the CD33-related subgroup of Siglecs and displays evolutionary variation among different mammalian species.Citation53 Its presence in microglia within the brain is particularly intriguing due to its association with AD susceptibility.Citation54
NLRP3 Inflammasome
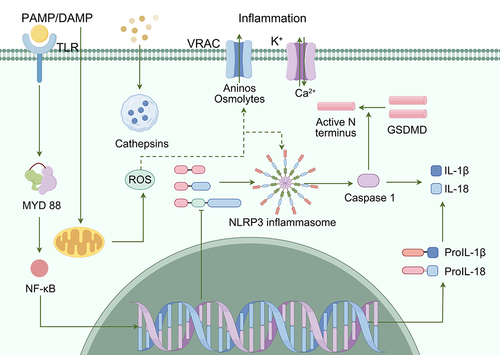
Nucleotide-binding domain-like receptor protein 3 (NLRP3) is widely distributed in the CNS and is highly expressed in microglia. Microglia are among the major regulators of the neuroinflammatory response, and neurodegeneration involves overactivation of microglia and upregulation of proinflammatory factors, which play important roles in neurodegenerative diseases.Citation55–57 In addition, abnormal Aβ amyloid aggregation, mitochondrial dysfunction, autophagy, and defective dopamine receptor function are strongly associated with microglial NLRP3 inflammasome-mediated neuroinflammation.Citation58–60
The NLRP3 inflammasome is a multiprotein complex composed of the pattern recognition receptor NLRP3, the junction protein apoptosis-associated speck-like protein, and the aspartate-specific cysteine protease 1 precursor.Citation61 The NLRP3 receptor is expressed in glial cells and the CNS and is composed of three distinct structural domains: the leucine-rich repeat (LRR) domain, the nucleoside triphosphatase domain, and the pyrin domain (PYD).Citation61 The LRR domain recognizes PAMPs or DAMPs, the nucleoside triphosphatase domain plays a crucial role in receptor activation, and the PYD domain mediates downstream signaling. When cells are exposed to DAMP- or PAMP-associated stimuli, which are recognized by the LRR at the carboxyl terminus of NLRP3, NLRP3 oligomerizes and accumulates the splice protein apoptosis-associated speck-like protein through PYD-PYD interactions and then binds to the pro-caspase-1 domain to assemble the inflammasome complex, which induces Caspase-1 autocatalysis.Citation62,Citation63 Finally, inflammatory factors such as interleukin (IL)-1β and IL-18 are cleaved from inactive precursors to activated forms and released from the cell.Citation56,Citation57 Additionally, activated Caspase-1 can trigger pyroptosis, a form of inflammatory programmed cell death characterized by plasma membrane rupture, the release of inflammatory mediators, and DNA damage.Citation64,Citation65 The mechanism of pyroptosis remains unclear.Citation66 Activated Caspase-1 cleaves the GSDMD protein into two parts (the N-terminal domain and C-terminal domain) by inducing enzymatic activity. The N-terminal domain of GSDMD can form pores in the lipid membrane, destroy the osmotic pressure balance between the inside and outside of the cell, cause water influx and membrane expansion, ultimately leading to membrane dissolution and inducing the apoptosis of microglia and neuronsCitation67,Citation68 ().
Figure 5 Schematic diagram of NLRP3 inflammasome signaling pathway.
TREM2 and AD
In the pathogenesis of AD, the accumulation of Aβ in the brain is often accompanied by increased glial cell proliferation and lipid deposition, leading to the progressive development of neuroinflammation.Citation69 Microglia can recognize amyloid through receptors such as TREM2, Toll-like receptors, and CR1, triggering a signaling cascade that prompts microglia to detect and migrate to the site of injury.Citation70–72 Subsequently, soluble Aβ and tau proteins are engulfed by microglia through phagocytosis, and upon fusion with lysosomes, vesicles and their contents are released and degraded,Citation73 ultimately reducing Aβ levels and slowing the formation of senile plaques. This process also helps prevent the formation of NFTs, thereby aiding in the prevention and delay of AD onset and clinical progression.Citation74 Senile plaques are nerve spots formed by abnormal deposition of Aβ39-42 in the brain. Aβ39-42 is a protein composed of 39~42 amino acid residues formed by amyloid precursor protein (APP) cleaved by β- and γ-secretory enzymes. Aβ exists in monomeric, oligomeric, and fibrillar forms, with oligomeric Aβ being the primary contributor to cognitive impairment and neurodegeneration in AD. Aβ oligomers can bind to the following receptors and influence nerve cell function through distinct signaling pathways.Citation75 Research has revealed that microglia rich in TREM2 play a crucial role in surrounding and compacting early amyloid fibrils and plaques. In mice lacking TREM2 or DAP12, as well as in individuals with the R47H mutation in the TREM2 gene, microglia exhibit a significantly diminished capacity to encase amyloid deposits.Citation76
Consequently, there was an increase in the formation of more diffuse plaques containing elongated and branched amyloid fibrils, leading to increased exposure of neighboring neurites.Citation76 This alteration was accompanied by intensified neuritic tau hyperphosphorylation and axonal degeneration surrounding amyloid deposits.Citation76 The extracellular domain of TREM2 is responsive to the accumulation of Aβ in AD,Citation77 and TREM2 has the ability to bind to various forms of Aβ, with the strongest affinity for Aβ oligomers.Citation78 Knocking out TREM2 results in impaired proliferation and activation of microglia near the lesion site, preventing them from carrying out their normal brain-protective functions effectively and ultimately worsening disease progression.Citation79–81 In mouse models of AD, microglia lacking TREM2 are unable to multiply and form clusters around the Aβ plaques associated with AD.Citation36 TREM2‐deficient N9 microglial cell lines, macrophages, and primary microglia all exhibited significantly decreased uptake of antibody‐bound Aβ, leading to reduced clearance of amyloid plaques.Citation82 This study revealed a decrease in the expression of APOE and APOC1 in TREM2-deficient human microglia. Differentially expressed genes were found to be enriched in pathways related to “calcium signaling pathway regulation”, “ERK1 and ERK2 cascade”, and “cell migration”. Upon activation of the TREM2 pathway, downregulated genes in TREM2-deficient microglia were significantly upregulated, leading to a shift in the enrichment pathway toward “positive regulation of leukocyte chemotaxis”, “cellular response to tumor necrosis factor”, and “positive regulation of the ERK1 and ERK2 cascade”.Citation83 In terms of metabolism, TREM2 plays a role in maintaining cellular energy and biosynthesis, contributing to the pathological process of AD.Citation71 Additionally, an increase in the number of autophagic vesicles was detected in the microglia of a TREM2-knockout 5XFAD mouse model, which was reduced upon the addition of cyclocreatine to improve energy storage, indicating energy metabolism disorders in the microglia of TREM2-knockout mice.Citation71 In terms of cell activity, a study showed that TREM2 enhances microglial survival through the activation of the Wnt/β-catenin signaling pathway. This finding also suggested that restoring Wnt/β-catenin signaling can be achieved when TREM2 activity is inhibited or decreased.Citation84 This highlights the potential of targeting the TREM2/β-catenin signaling pathway for AD treatment. Research has identified a group of lipoprotein particles, including LDL, and apolipoproteins, such as CLU/APOJ and APOE, as ligands of TREM2.Citation82 The binding of these ligands by TREM2 is hindered or decreased by disease-associated mutations such as AD. Aβ attaches to lipoproteins, and this combination is effectively absorbed by microglia in a TREM2-dependent manner.Citation82 Microglia lacking Trem2 displayed decreased internalization of LDL and CLU. The absorption of Aβ-lipoprotein complexes was diminished in macrophages from individuals with a TREM2 AD variant.Citation82 These findings establish a connection between three genetic risk factors for AD and shed light on a potential mechanism through which mutant TREM2 increases the risk of AD.
Microglia play a crucial role in controlling synaptic remodeling within the CNS. Activation of the classical complement pathway facilitates microglia-driven synaptic pruning both in developmental stages and during disease progression.Citation85 Studies using conditional knockout mice revealed that the specific deletion of SIRPα in microglia leads to a reduction in synaptic density.Citation85 In human tissue, a decrease in microglial SIRPα expression has been noted to be correlated with the progression of AD.Citation85 CD47, which shields synapses from excessive pruning during development, points to the involvement of microglial SIRPα, a receptor for CD47, in the process of synaptic remodeling.Citation85,Citation86 Furthermore, the absence of microglial SIRPα results in an increased loss of synapses due to microglial engulfment, ultimately leading to heightened cognitive impairment.Citation85,Citation87 A separate study revealed that the extracellular domain of TREM2 binds strongly to C1q, which is responsible for initiating the classical complement pathway, effectively inhibiting the activation of this pathway.Citation88,Citation89 In postmortem brain tissue samples from AD patients, TREM2 was observed to form protein complexes with C1q. The number of these complexes was found to have a negative correlation with the level of complement protein C3 deposition and a positive correlation with synaptic protein levels.Citation88 Using a mouse model of AD in which the Trem2 gene was absent, researchers noted that in the early stages of the disease, the absence of the Trem2 gene did not impact the level of AD pathologic protein deposition or the morphology or quantity of microglia.Citation88 However, in a mouse model of AD in which the Trem2 gene was absent, researchers noted that in the early stages of the disease, the absence of the Trem2 gene did not impact the level of AD pathologic protein deposition or the morphology or quantity of microgliaCitation88 ().
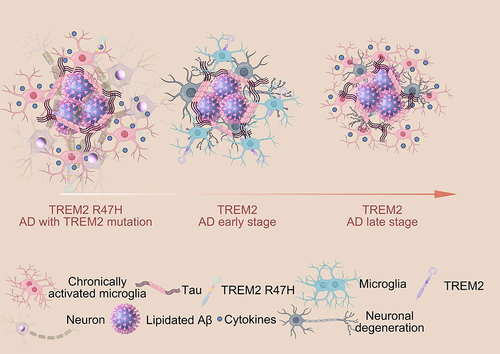
Figure 6 Function of TREM2 in AD.Citation90 (TREM2 expression plays a crucial role in regulating the proliferation, migration, and phagocytosis ability of microglia. In the early stages of AD, microglia expressing TREM2 effectively surround and interact with lipidated Aβ plaques to clear them, thus preventing the spread of Aβ. However, in the late stages of AD, these TREM2-expressing microglia struggle to clear the aggregates, leading to chronic inflammation, reduced phagocytosis ability, and the induction of tau phosphorylation and aggregation. Individuals with TREM2 mutations experience weakened TREM2 affinity, resulting in impaired microglial proliferation and migration, which hinders the effective clearance of Aβ aggregates. Furthermore, TREM2 mutations lead to increased cytokine secretion from microglia, ultimately exacerbating the spread of Aβ and the phosphorylation and aggregation of tau, thereby worsening AD pathology).
sTREM2 and AD
sTREM2 can penetrate the cerebrospinal fluid-brain barrier and is found at heightened levels in the cerebrospinal fluid of patients with various neurological disorders, including AD, Parkinson’s disease, frontotemporal dementia with granulosomal protein mutations, and individuals with normal cognitive function who are undergoing natural aging.Citation91–94 This finding suggested that sTREM2 levels can serve as an indicator of microglial activation status and neuronal degeneration.Citation95 Research has validated sTREM2 as a potential biomarker for detecting AD.Citation96 Moreover, no vesicular phagocytosis of APOE was detected in TREM2-deficient microglia.Citation97 Furthermore, studies have revealed that sTREM2 may play a role in reducing microglial apoptosis by activating the Akt-GSK3β-β-catenin signaling pathway and promoting microglial survival through the PI3K/Akt signaling pathway, thereby exerting neuroprotective effects.Citation39,Citation83 Experiments in which the sTREM2-Fc protein was injected into the hippocampus of normal mice and TREM2-knockout mice revealed increased expression levels of inflammatory factors, altered microglial morphology, and enhanced immune responses, suggesting that enhancing the sTREM2 signaling pathway could be a therapeutic approach for AD.Citation98 In addition, direct injection of recombinant sTREM2 protein or adenovirus transfection into 5xFAD mice to increase sTREM2 levels can promote the proliferation and phagocytic activity of microglia near amyloid plaques and accelerate the phagocytosis and clearance of Aβ plaques.Citation98 One study was the first to examine CSF sTREM2 within the amyloid/tau/neurodegeneration classification framework. The findings from the AD Imaging Initiative cohort show that elevated levels of CSF sTREM2 in the early stages correspond with increased tau pathology and neurodegeneration. Conversely, lower levels of CSF sTREM2 are linked to Aβ deposition without tau deposition and neurodegeneration. Studies have shown that microglial phagocytosis may not always be beneficial, and its effects vary depending on the specific clinical stage. In patients with AD who exhibit only pathophysiological changes without significant clinical symptoms, increased microglial phagocytosis can decrease Aβ levels and slow the formation of SPs and NFTs.Citation74 However, due to the limited number of lysosomes, microglial phagocytosis decreases after the uptake of Aβ and tau proteins.Citation99 Subsequent to phagocytosis, the inflammatory vesicular cascade triggered by cellular metabolism, including NLRP3 and inflammatory cytokines such as TNF-α and IL-1, leads to microglial polarization toward a proinflammatory state. This, in turn, induces the secretion of proinflammatory cytokines, exacerbates the inflammatory response, hampers Aβ phagocytosis, and promotes the pathological accumulation of the Tau protein.Citation100,Citation101
CD33 and AD
The number of CD33-expressing microglia was found to be positively correlated with plaque burden and cognitive decline in AD.Citation102,Citation103 However, higher CD33 expression in AD may be a response to disease pathology, such as brain inflammation. Research has shown that specific cytokines, such as IL-15, can induce the upregulation of CD33.Citation104,Citation105 CD33 is predominantly expressed by microglia in the brain, suggesting that the genetic correlation between CD33 and AD susceptibility may be due to the regulation of microglial cell function by CD33.Citation106,Citation107 Although a contributing role in myeloid cells outside the brain cannot be definitively ruled out, initial findings indicated increased expression of CD33 on microglia in AD brains.
The connection between CD33 and AD was initially revealed through genome-wide association studies, which identified the rs3826656 SNP as a susceptibility factor for late-onset AD.Citation45 Studies have shown increased CD33 expression in microglia within the AD brain, and the protective allele of the CD33 SNP rs3865444 was found to be correlated with decreased CD33 expression and insoluble Aβ42 levels in the brain.Citation102 The rs3865444 SNP is located in the CD33 promoter, which is located 372 base pairs upstream of the transcription start site. Initially, thought to affect CD33 gene expression,Citation108 further analysis revealed a coinherited SNP, rs12459419, located four nucleotides into exon 2 of CD33, which influences mRNA splicing.Citation108,Citation109 A recently identified CD33 SNP, rs2455069, has been suggested to be linked to the risk of developing AD in a small group of Italian patients.Citation110 In silico analysis indicated that a change in the amino acid at position 69 of hCD33 from arginine to glycine in the rare rs2455069 SNP could increase the affinity for Sia-containing ligands.Citation110 Further investigation is needed to confirm the association of this SNP with AD susceptibility in a larger patient population.
In the context of developing novel and effective approaches for treating AD, immunomodulatory receptors on microglia, including CD33, are considered promising targets for drug discovery.Citation111 Notably, various anti-CD33 antibody targeting strategies have made progress in the clinical treatment of acute myeloid leukemia.Citation111,Citation112 CD33 has been found to hinder microglial uptake and clearance of Aβ42, leading to a reduced plaque burden in APP/PS1 CD33-/-mice.Citation102 Higher levels of CD33 expression in the brain are associated with greater cognitive declineCitation103 and increased AD pathology,Citation75 making CD33 a potential therapeutic target for AD. Research has shown that CD33 knockout in 5xFAD mice leads to a decrease in Aβ pathology and an improvement in cognition.Citation113 However, the effects of TREM2 knockout were reversed.Citation113 TREM2 knockout resulted in a reduction in the clustering of Iba1+ myeloid cells around plaques, a phenomenon that could not be reversed by CD33 knockout.Citation113 Furthermore, differential gene expression in 5xFAD; CD33−/− microglia was found to be dependent on the presence of TREM2.Citation113 These findings suggest that TREM2 functions downstream of CD33 and that the loss of microglial clearance capacity could be reversed through therapeutic inhibition of CD33.Citation114
The application of antibodies remains an exciting therapeutic option for the treatment of human diseases. With features such as high target specificity, slow hepatic or renal metabolism, and the need for less frequent administration, antibodies are attractive small molecules.Citation115 Anti-CD33 antibodies have a long history of use in the clinic for treating leukemia,Citation116–118 with gemtuzumab ozogamicin being the first approved antibody‒drug conjugate for treating acute myeloid leukemia.Citation119 These antibodies target the V-set domain (encoded by exon 2), exclusively targeting hCD33M, and show promise for preclinical studies in AD patients.Citation118,Citation120 A phase 1b clinical trial of AL003, a human-specific anti-CD33 monoclonal antibody developed for AD treatment, was successfully completed. Promising assessments of tolerability and pharmacokinetics have been reported, with the antibody being well tolerated and showing target engagement in peripheral and CNS compartments.Citation111 As a result, AL003 is now being considered for further investigation in a proof-of-concept Phase 2 study.Citation111
NLRP3 Inflammatory Complex and AD
Numerous studies have demonstrated the involvement of the NLRP3 inflammasome in regulating the neuroinflammatory response in AD, highlighting its critical role in the pathogenesis and progression of AD.Citation121,Citation122 Indeed, Aβ activation of the NLRP3 inflammasome in microglia is fundamental for IL-1β maturation and subsequent inflammatory events.Citation123,Citation124 In 2008, HalleCitation123 et al reported that the injection of Aβ into the lateral ventricle of mice could activate the NLRP3 inflammasome in microglia, causing the maturation and secretion of IL-1β and IL-18 mediated by Caspase-1, which was the first direct demonstration of the role of the NLRP3 inflammasome in AD. Further studies showed that knocking out the NLRP3 gene in APP/PS1 transgenic mice reduced caspase-1 activity, enhanced Aβ phagocytosis by microglia, and decreased Aβ deposition.Citation125 Additionally, spatial memory deficits in APP/PS1 mice were mitigated. These findings are supported by other researchers, indicating the important role of the NLRP3 inflammasome in AD.Citation126–128 HenekaCitation125 reported that mice with mutations linked to familial AD, specifically Nlrp3−/− or Casp1−/−, exhibited significant protection against spatial memory loss and other symptoms of AD. These mice exhibited decreased activation of brain caspase-1 and IL-1β, along with improved clearance of amyloid-β. Additionally, the absence of the NLRP3 inflammasome led to a shift in microglia toward the M2 phenotype and reduced Aβ deposition in the APP/PS1 model of AD.Citation125 These findings highlight the importance of the NLRP3/caspase-1 axis in AD pathogenesis and suggest that inhibiting the NLRP3 inflammasome is a promising therapeutic approach for this condition.
The activation products of the NLRP3 inflammasome, IL-1β and IL-18, are crucial for initiating and perpetuating the neuroinflammatory response in AD.Citation129,Citation130 IL-1β and IL-18 produced after inflammasome activation recruit intracellular junction molecules (MyD88, IRAK and TRAFl5) through their respective receptors, IL-1βr and IL-18R, to activate NF-κB and the c-Jun N-terminal kinase, and the p38 MAPK signaling pathway simultaneously activates a variety of inflammatory cells, such as microglia and astrocytes, inducing them to transform into proinflammatory phenotypes, secrete more inflammatory factors and proinflammatory mediators, such as IL-1β, IL-6, IL-18 and cyclooxygenase-2, macrophage inflammatory proteins (MIP-1α, MIP-1β and MIP-2) and inducible nitric oxide synthase,Citation130 induce inflammatory cascade reactions and aggravate AD. A substantial body of evidence indicates that targeting the glia–neuron cycle could be a promising strategy for developing new therapeutic approaches for AD that could alter the progression of the disease.Citation131–134 Research strongly suggests that IL-1 plays a crucial role in the pathogenesis and progression of AD.Citation135,Citation136 Analysis of AD brain tissue revealed an overproduction of IL-1, particularly in activated microglia surrounding Aβ plaques and neurons with NFTs, the two main neuropathological features of AD.Citation137 This overproduction of IL-1 is closely associated with the severity of neuropathology in a specific brain region.Citation137 Additionally, studies on cells have demonstrated that IL-1 can induce the production of various harmful molecules from microglia, astrocytes, and neurons. For instance, IL-1 can trigger the production of α-1 anti-chymotrypsin, IL-6, S100B, and inducible nitric oxide synthase, which are elevated in the AD brain. These molecules, either independently or by promoting the production of other molecules, contribute to a neuroinflammatory cascade that is believed to lead to cell damage, dysfunction, and death in AD.Citation138 This theory is reinforced by the observed neuroprotection when the neuroinflammatory cascade is suppressed in AD animal models. Furthermore, several studies on IL-1 genetics have revealed that polymorphisms in the IL-1 and IL-1 receptor genes can increase the risk of AD by up to three times in homozygous carriers.Citation139 Multiple studies have indicated that IL-1β can have a detrimental effect on hippocampus-dependent learning and may inhibit long-term potentiation, with the extent of impact varying based on concentration.Citation140 Additionally, other inflammatory cytokines produced by microglia have been shown to similarly decrease long-term potentiation.Citation141 Chronic administration of an IL-1R blocking antibody to 3xTg-AD mice results in significant changes in brain inflammatory responses, improvements in cognitive deficits, reductions in tau pathology, and partial decreases in certain forms of Aβ.Citation142 These alterations in inflammatory responses are associated with decreased NF-κB activity.Citation142 Additionally, blocking IL-1 signaling leads to reduced activity of various tau kinases in the brain, such as cdk5/p25, glycogen synthase kinase 3β, and p38-MAPK, as well as decreased levels of phosphorylated tau.Citation142 One study also revealed a decrease in the levels of the astrocyte-derived cytokine S100B and neuronal Wnt/β-catenin signaling in 3xTg-AD brains.Citation142 The proinflammatory factor IL-18 exhibited a similar impact to that of IL-1β in AD. In vitro experiments revealed that IL-18 enhances Aβ deposition in human neuroblastoma SHSY5Y cells, which are commonly used as a neuronal model.Citation143 Furthermore, IL-18 was shown to upregulate the expression of glycogen synthase kinase 3β and cyclin-dependent kinase 5, both of which are implicated in the hyperphosphorylation of the Tau protein.Citation144 Studies have indicated that IL-18 may induce the protein expression of APP, β-site APP-Cleenzyme 1, and certain subunits of the γ-secretase complex, potentially accelerating Aβ production.Citation145 Moreover, meta-analyses have linked gene polymorphisms in the IL-18 promoter region to an increased risk and poorer prognosis of AD.Citation146 These findings suggest that immune mechanisms mediated by IL-1β and IL-18, which are products of the NLRP3 inflammatory complex, play crucial roles in the pathological progression of AD ().
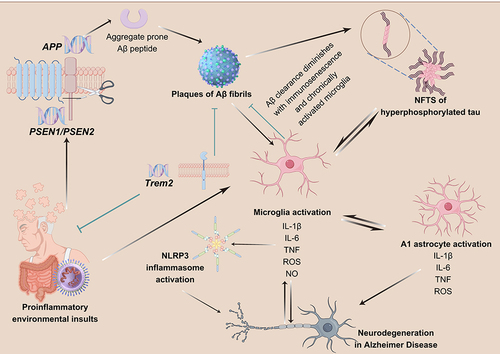
Figure 7 The role of NLRP3 inflammasome in AD.Citation147 (Microglia are activated in response to DAMPs from Aβ plaques, and are mobilized to eliminate them. However, prolonged activation of microglia and immunosenescence can reduce their effectiveness over time. TREM2 plays a role in regulating the activity and survival of microglia. Mutations in TREM2 can affect the ability of microglia to regulate cytokine production and clear neural debris. Failure to clear Aβ plaques can lead to the formation of NFTs, triggered by both the plaques and the persistently activated microglia. Additionally, hyperphosphorylated tau protein can further stimulate microglia.
Discussion
The development of drugs for AD has progressed slowly, and the currently approved drugs can only improve cognitive symptoms for a limited time and cannot reverse the disease process. Various interventions targeting Aβ amyloid deposition, tau hyperphosphorylation, neuroinflammation, and neuroprotection are under investigation.Citation148,Citation149 In recent years, most studies on the role of neuroinflammation in the development of AD have focused on microglia, astrocytes and neurons. Targeted therapy for these cells within the CNS or reducing CNS inflammation by impacting peripheral inflammatory processes could offer a new approach for diagnosing and treating AD. At present, the number of trials focusing on anti-neuroinflammatory drugs in Phase I and Phase II trials has notably increased. Drug experimental studies on microglia are still in the early stages, with a primary focus on regulating the phenotype of microglia. Study has found that promoting a microglial switch from the inflammatory M1 phenotype to the protective M2 phenotype in APP/PS1 mice can have a neuroprotective effect and improve cognitive dysfunction in mice with AD.Citation150 Another research has indicated that peroxisome proliferator-activated receptor gamma (PPAP-γ) agonists can elevate M2 phenotype microglia markers such as CD206, IL-4, TGF-β, G-CSF levels, while reducing the levels of M1 phenotype microglia markers like CD86, COX-2, iNOS, IL-1β, and IL-6. This demonstrates their ability to shift microglial cells from an M1 phenotype to an M2 phenotype, enhancing their phagocytic function and promoting amyloid clearance in animal models. However, there is no evidence supporting the efficacy of Rosiglitazone Monotherapy(PPAP-γ agonists) in improving cognition or global function in APOE-Ε4-negative individuals or other analysis populations.Citation151 The findings suggest that there is still significant potential for further exploration in modulating microglia for the treatment of AD. However, further verification in vivo, particularly in human studies, is necessary.
In a meta-analysis of 40 studies on blood and 14 on CSF, individuals with AD exhibited elevated levels of IL-6, TNF-α, IL-1β, TGF-β, IL-12, and IL-18 in blood, as well as increased levels of TGF-β in CSF, when compared to control groups.Citation151,Citation152 A more recent meta-analysis of 175 blood studies revealed heightened levels of IL-1β, IL-2, IL-6, IL-18, IFN-γ, TNF-α converting enzyme, soluble TNF receptors 1 and 2, and reduced levels of IL-1 receptor antagonist and leptin in AD patients compared to controls.Citation153 These findings provide further support for the presence of inflammatory responses in individuals with AD. Candidate markers associated with microglia have been extensively investigated. One study found that YKL-40, a protein primarily produced by astrocytes and encoded by the Chi3l1 gene, is a well-researched biomarker in CSF that tends to increase with age and in the early stages of ADCitation154 and in the late preclinical AD stages compared with early preclinical stages.Citation155 Using a mouse model of AD (APP/PS1), researchers observed that deleting Chi3l1 led to a reduction in Aβ accumulation and an increase in the expression of the microglial lysosomal marker CD68 around the plaques.Citation154 This suggests that Chi3l1 may inhibit the activation of glial cells involved in phagocytosis and contribute to the buildup of amyloid. In line with this, lowering Chi3l1 levels was found to enhance the phagocytosis of zymosan particles and Aβ peptide by both astrocytes and microglia in vitro.Citation154 Activated microglia express translocator protein 18 kDa (TSPO) on the outer membrane of their mitochondria. TSPO contains an isoquinoline site that binds 11C-(R)-PK11195. TSPO PET imaging has revealed varying degrees of microglial activation in groups of patients with clinically probable AD and cases of amnestic mild cognitive impairment (MCI).Citation156 A study has provided evidence supporting the role of neuroinflammation in the neurodegenerative pathology of a majority of MCI cases related to AD.Citation157 Statistical parametric mapping identified clusters of elevated PK11195 binding in 85% of our Aβ-positive amnestic MCI (prodromal AD) subjects.Citation157 The aforementioned research offers a novel approach to the diagnosis and intervention of AD through microglia.
While microglia and neuritis are recognized as crucial components in the pathological mechanisms of AD, the role of microglia in this process is multifaceted, encompassing both neuroprotective and neurotoxic functions, akin to a double-edged sword. The intricate actions of microglia in AD are predominantly influenced by TREM2 and its regulation of associated genes such as CD33 and NLRP3. Consequently, targeting the inflammasome of TREM2 and NLRP3 holds promise as a therapeutic strategy for treating AD in the future. Nevertheless, it is imperative to acknowledge that the precise mechanisms by which the NLRP3 inflammasome contributes to the onset and progression of AD, as well as the upstream regulatory mechanisms governing NLRP3 inflammasome activation, necessitate further elucidation. Additionally, the TREM2-APOE pathway has emerged as a critical modulator of the functional phenotype of microglia in AD, suggesting that this pathway is a potential novel target for restoring microglial homeostasis.Citation158 Hence, delving deeper into this research direction is likely to reveal additional therapeutic strategies for AD and present novel interventions for its clinical management, thereby offering fresh perspectives on the treatment of this debilitating condition.
Conclusion
The role of microglia in AD is intricate, demonstrating both neuroprotective and neurotoxic effects, with its actions largely influenced by the regulation of other related genes. Targeting microglia is a promising approach to delaying AD progression; however, challenges such as drug bioavailability, specificity, and safety remain unresolved. Enhancing the targeting specificity of microglia could offer novel intervention strategies for treating AD in humans.
Abbreviations
AD, Alzheimer’s disease; CNS, Central nervous system; Aβ, Amyloid-β; NFTs, Neurofibrillary tangles; TREM2, Triggering receptor expressed on myeloid cells-2; APOE, Apolipoprotein E; PGRN, Progranulin; CV, Common variant; PAMPs, Pathogen-associated molecular patterns; DAMPs, Damage-associated molecular patterns; sTREM2, soluble Triggering receptor expressed on myeloid cells-2; PI3K, Phosphatidylinositide 3-kinases; mTOR, mammalian Target of rapamycin; MAPK, Mitogen-activated protein kinase; NLRP3, Nucleotide-binding domain-like receptor protein 3; LRR, Leucine-rich repeat; PYD, Pyrin domain; IL, Interleukin; APP, Amyloid precursor protein; TSPO, Translocator protein of 18KD; MCI, Mild cognitive impairment.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
Acknowledgments
The images in this paper are courtesy of Figdraw (www.figdraw.com) for providing technical support and materials.
Additional information
Funding
References
- Yan H, Feng L, Li M. The role of traditional Chinese medicine natural products in beta-Amyloid deposition and tau protein hyperphosphorylation in Alzheimer’s disease. Drug Des Devel Ther. 2023;17:3295–3323. doi:10.2147/DDDT.S380612
- Azarpazhooh MR, Avan A, Cipriano LE, et al. A third of community-dwelling elderly with intermediate and high level of Alzheimer’s neuropathologic changes are not demented: a meta-analysis. Ageing Res Rev. 2020;58(101002):101002. doi:10.1016/j.arr.2019.101002
- Penney J, Ralvenius WT, Tsai LH. Modeling Alzheimer’s disease with iPSC-derived brain cells. Mol Psychiatry. 2020;25(1):148–167. doi:10.1038/s41380-019-0468-3
- Jiang L, Xu L, Meng X. Recent advance in role of immune system in pathogenesis of Alzheimer’s disease and its immunotherapies. Chin J Neuromed. 2022;(02):200–206. doi:10.3760/cma.j.cn115354-20210925-00627
- Nizami S, Hall-Roberts H, Warrier S, Cowley SA, Di Daniel E. Microglial inflammation and phagocytosis in Alzheimer’s disease: potential therapeutic targets. Br J Pharmacol. 2019;176(18):3515–3532. doi:10.1111/bph.14618
- Herrup K. The case for rejecting the amyloid cascade hypothesis. Nat Neurosci. 2015;18(6):794–799. doi:10.1038/nn.4017
- Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol. 2021;17(3):157–172. doi:10.1038/s41582-020-00435-y
- Calsolaro V, Edison P. Neuroinflammation in Alzheimer’s disease: current evidence and future directions. Alzheimers Dement. 2016;12(6):719–732. doi:10.1016/j.jalz.2016.02.010
- Villegas-Llerena C, Phillips A, Garcia-Reitboeck P, Hardy J, Pocock JM. Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr Opin Neurobiol. 2016;36:74–81. doi:10.1016/j.conb.2015.10.004
- Wang Q, Sun J, Chen T, et al. Ferroptosis, pyroptosis, and cuproptosis in Alzheimer’s disease. ACS Chem Neurosci. 2023;14(19):3564–3587. doi:10.1021/acschemneuro.3c00343
- Mangalmurti A, Lukens JR. How neurons die in Alzheimer’s disease: implications for neuroinflammation. Curr Opin Neurobiol. 2022;75:102575. doi:10.1016/j.conb.2022.102575
- Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci. 2018;21(10):1359–1369. doi:10.1038/s41593-018-0242-x
- Salter M, Stevens W. B. Microglia emerge as central players in brain disease. Nat Med. 2017;23(9):1018–1027. doi:10.1038/nm.4397
- Ueno M, Fujita Y, Tanaka T, et al. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci. 2013;16(5):543–551. doi:10.1038/nn.3358
- Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp Med. 2005;201(4):647–657. doi:10.1084/jem.20041611
- Roumier A, Béchade C, Poncer JC, et al. Impaired synaptic function in the microglial KARAP/DAP12-deficient mouse. J Neurosci. 2004;24(50):11421–11428. doi:10.1523/JNEUROSCI.2251-04.2004
- Ji K, Akgul G, Wollmuth LP, Tsirka S. E. Microglia actively regulate the number of functional synapses. PLoS One. 2013;8(2):e56293. doi:10.1371/journal.pone.0056293
- Jessen KR. Glial cells. Int J Biochem Cell Biol. 2004;36(10):1861–1867. doi:10.1016/j.biocel.2004.02.023
- Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. 2017;35:441–468. doi:10.1146/annurev-immunol-051116-052358
- Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–845. doi:10.1126/science.1194637
- Baufeld C, O’Loughlin E, Calcagno N, Madore C, Butovsky O. Differential contribution of microglia and monocytes in neurodegenerative diseases. J Neural Transm. 2018;125(5):809–826. doi:10.1007/s00702-017-1795-7
- Heneka MT, Carson MJ, El KJ, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388–405. doi:10.1016/S1474-4422(15)70016-5
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140(6):918–934. doi:10.1016/j.cell.2010.02.016
- Luo XG, Chen SD. The changing phenotype of microglia from homeostasis to disease. Transl Neurodegener. 2012;1(1):9. doi:10.1186/2047-9158-1-9
- Subhramanyam CS, Wang C, Hu Q, Dheen ST. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin Cell Dev Biol. 2019;94:112–120. doi:10.1016/j.semcdb.2019.05.004
- Jay TR, Hirsch AM, Broihier ML, et al. Disease Progression-Dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J Neurosci. 2017;37(3):637–647. doi:10.1523/JNEUROSCI.2110-16.2016
- Ulrich JD, Ulland TK, Mahan TE, et al. ApoE facilitates the microglial response to amyloid plaque pathology. J Exp Med. 2018;215(4):1047–1058. doi:10.1084/jem.20171265
- Saeedi-Boroujeni A, Purrahman D, Shojaeian A, Poniatowski LA, Rafiee F, Mahmoudian-Sani MR. Progranulin (PGRN) as a regulator of inflammation and a critical factor in the immunopathogenesis of cardiovascular diseases. J Inflamm. 2023;20(1):1. doi:10.1186/s12950-023-00327-0
- Xu W, Tan CC, Cao XP, Tan L. Neuroinflammation modulates the association of PGRN with cerebral amyloid-beta burden. Neurobiol Aging. 2021;103:52–59. doi:10.1016/j.neurobiolaging.2021.02.016
- Xu W, Han SD, Zhang C, et al. The FAM171A2 gene is a key regulator of progranulin expression and modifies the risk of multiple neurodegenerative diseases. Sci Adv. 2020;6(43). doi:10.1126/sciadv.abb3063
- Zhou X, Sun L, Brady OA, Murphy KA, Hu F. Elevated TMEM106B levels exaggerate lipofuscin accumulation and lysosomal dysfunction in aged mice with progranulin deficiency. Acta Neuropathol Commun. 2017;5(1):9. doi:10.1186/s40478-017-0412-1
- Van Kampen JM, Baranowski D, Kay DG. Progranulin gene delivery protects dopaminergic neurons in a mouse model of Parkinson’s disease. PLoS One. 2014;9(5):e97032. doi:10.1371/journal.pone.0097032
- Minami SS, Min SW, Krabbe G, et al. Progranulin protects against amyloid beta deposition and toxicity in Alzheimer’s disease mouse models. Nat Med. 2014;20(10):1157–1164. doi:10.1038/nm.3672
- Condello C, Yuan P, Schain A, Grutzendler J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Abeta42 hotspots around plaques. Nat Commun. 2015;6:6176. doi:10.1038/ncomms7176
- Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer’s disease. J Cell Biol. 2018;217(2):459–472. doi:10.1083/jcb.201709069
- Song WM, Joshita S, Zhou Y, Ulland TK, Gilfillan S, Colonna M. Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism. J Exp Med. 2018;215(3):745–760. doi:10.1084/jem.20171529
- Li X, Ni S, Yao Z, Zhang Q. Research progress of TREM2 in Alzheimer’s disease. Chinese Pharmacological Bulletin. 2020;36(08):1049–1053. doi:10.3969/j.issn.1001-1978.2020.08.004
- Berner DK, Wessolowski L, Armbrust F, et al. Meprin beta cleaves TREM2 and controls its phagocytic activity on macrophages. FASEB J. 2020;34(5):6675–6687. doi:10.1096/fj.201902183R
- Zhong L, Chen XF, Wang T, et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J Exp Med. 2017;214(3):597–607. doi:10.1084/jem.20160844
- Konishi H, Kiyama H. Microglial TREM2/DAP12 signaling: a Double-Edged sword in neural diseases. Front Cell Neurosci. 2018;12(206). doi:10.3389/fncel.2018.00206
- Ulrich JD, Ulland TK, Colonna M, Holtzman DM. Elucidating the role of TREM2 in Alzheimer’s disease. Neuron. 2017;94(2):237–248. doi:10.1016/j.neuron.2017.02.042
- Wang Y, Cella M, Mallinson K, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. 2015;160(6):1061–1071. doi:10.1016/j.cell.2015.01.049
- Mazaheri F, Snaidero N, Kleinberger G, et al. TREM2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep. 2017;18(7):1186–1198. doi:10.15252/embr.201743922
- Efthymiou AG, Goate AM. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol Neurodegener. 2017;12(1):43. doi:10.1186/s13024-017-0184-x
- Bertram L, Lange C, Mullin K, et al. Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am J Hum Genet. 2008;83(5):623–632. doi:10.1016/j.ajhg.2008.10.008
- Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368(2):117–127. doi:10.1056/NEJMoa1211851
- Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43(5):429–435. doi:10.1038/ng.803
- Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43(5):436–441. doi:10.1038/ng.801
- Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368(2):107–116. doi:10.1056/NEJMoa1211103
- Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol. 2007;7(4):255–266. doi:10.1038/nri2056
- Ishida A, Akita K, Mori Y, et al. Negative regulation of Toll-like receptor-4 signaling through the binding of glycosylphosphatidylinositol-anchored glycoprotein, CD14, with the sialic acid-binding lectin, CD33. J Biol Chem. 2014;289(36):25341–25350. doi:10.1074/jbc.M113.523480
- Linnartz-Gerlach B, Mathews M, Neumann H. Sensing the neuronal glycocalyx by glial sialic acid binding immunoglobulin-like lectins. Neuroscience. 2014;275:113–124. doi:10.1016/j.neuroscience.2014.05.061
- Cao H, Crocker PR. Evolution of CD33-related siglecs: regulating host immune functions and escaping pathogen exploitation? Immunology. 2011;132(1):18–26. doi:10.1111/j.1365-2567.2010.03368.x
- Miles LA, Hermans SJ, Crespi G, et al. Small molecule binding to Alzheimer risk factor CD33 promotes abeta phagocytosis. iScience. 2019;19:110–118. doi:10.1016/j.isci.2019.07.023
- Joers V, Tansey MG, Mulas G, Carta AR. Microglial phenotypes in Parkinson’s disease and animal models of the disease. Prog Neurobiol. 2017;155:57–75. doi:10.1016/j.pneurobio.2016.04.006
- Jose S, Groves NJ, Roper KE, Gordon R. Mechanisms of NLRP3 activation and pathology during neurodegeneration. Int J Biochem Cell Biol. 2022;151:106273. doi:10.1016/j.biocel.2022.106273
- Kwon HS, Koh SH. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl Neurodegener. 2020;9(1):42. doi:10.1186/s40035-020-00221-2
- Yan M, Jin H, Pan C, Han X. Chronic Microcystin-LR-Induced alpha-Synuclein promotes neuroinflammation through activation of the NLRP3 inflammasome in microglia. Mol Neurobiol. 2023;60(2):884–900. doi:10.1007/s12035-022-03134-5
- Lawrence G, Holley CL, Schroder K. Parkinson’s disease: connecting mitochondria to inflammasomes. Trends Immunol. 2022;43(11):877–885. doi:10.1016/j.it.2022.09.010
- Sun G, Zhang R, Liu C, Meng W, Pang Q. Itaconate attenuates neuroinflammation and exerts dopamine neuroprotection in Parkinson’s disease through inhibiting NLRP3 inflammasome. Brain Sci. 2022;12(9):1255. doi:10.3390/brainsci12091255
- Abderrazak A, Syrovets T, Couchie D, et al. NLRP3 inflammasome: from a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015;4:296–307. doi:10.1016/j.redox.2015.01.008
- Shao BZ, Xu ZQ, Han BZ, Su DF, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol. 2015;6(262). doi:10.3389/fphar.2015.00262
- Willingham SB, Allen IC, Bergstralh DT, et al. NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J Immunol. 2009;183(3):2008–2015. doi:10.4049/jimmunol.0900138
- Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821–832. doi:10.1016/j.cell.2010.01.040
- Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev. 2015;265(1):130–142. doi:10.1111/imr.12287
- Lechtenberg BC, Mace PD, Riedl SJ. Structural mechanisms in NLR inflammasome signaling. Curr Opin Struct Biol. 2014;29:17–25. doi:10.1016/j.sbi.2014.08.011
- Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006;8(11):1812–1825. doi:10.1111/j.1462-5822.2006.00751.x
- Ding J, Wang K, Liu W, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535(7610):111–116. doi:10.1038/nature18590
- Cai W, Wu T, Chen N. The Amyloid-Beta clearance: from molecular targets to glial and neural cells. Biomolecules. 2023;13(2). doi:10.3390/biom13020313
- Keren-Shaul H, Spinrad A, Weiner A, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169(7):1276–1290. doi:10.1016/j.cell.2017.05.018
- Ulland TK, Song WM, Huang SC, et al. TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell. 2017;170(4):649–663. doi:10.1016/j.cell.2017.07.023
- Das R, Chinnathambi S. Microglial remodeling of actin network by Tau oligomers, via G protein-coupled purinergic receptor, P2Y12R-driven chemotaxis. Traffic. 2021;22(5):153–170. doi:10.1111/tra.12784
- Doran AC, Yurdagul AJ, Tabas I. Efferocytosis in health and disease. Nat Rev Immunol. 2020;20(4):254–267. doi:10.1038/s41577-019-0240-6
- Kuruppu S, Rajapakse NW, Parkington HC, Smith AI. The characteristics of astrocyte on Abeta clearance altered in Alzheimer’s disease were reversed by anti-inflammatory agent (+)-2-(1-hydroxyl-4-oxocyclohexyl) ethyl caffeate. Am J Transl Res. 2017;9(7):3514–3516.
- Walker DG, Whetzel AM, Serrano G, Sue LI, Beach TG, Lue LF. Association of CD33 polymorphism rs3865444 with Alzheimer’s disease pathology and CD33 expression in human cerebral cortex. Neurobiol Aging. 2015;36(2):571–582. doi:10.1016/j.neurobiolaging.2014.09.023
- Yuan P, Condello C, Keene CD, et al. TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron. 2016;90(4):724–739. doi:10.1016/j.neuron.2016.05.003
- Zhao Y, Wu X, Li X, et al. TREM2 is a receptor for beta-Amyloid that mediates microglial function. Neuron. 2018;97(5):1023–1031. doi:10.1016/j.neuron.2018.01.031
- Lessard CB, Malnik SL, Zhou Y, et al. High-affinity interactions and signal transduction between Abeta oligomers and TREM2. EMBO Mol Med. 2018;10(11). doi:10.15252/emmm.201809027
- Wang S, Mustafa M, Yuede CM, et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J Exp Med. 2020;217(9). doi:10.1084/jem.20200785
- Ulrich JD, Finn MB, Wang Y, et al. Altered microglial response to Abeta plaques in APPPS1-21 mice heterozygous for TREM2. Mol Neurodegener. 2014;9:20. doi:10.1186/1750-1326-9-20
- Wang Y, Ulland TK, Ulrich JD, et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med. 2016;213(5):667–675. doi:10.1084/jem.20151948
- Xiang X, Werner G, Bohrmann B, et al. TREM2 deficiency reduces the efficacy of immunotherapeutic amyloid clearance. EMBO Mol Med. 2016;8(9):992–1004. doi:10.15252/emmm.201606370
- Mcquade A, Kang YJ, Hasselmann J, et al. Gene expression and functional deficits underlie TREM2-knockout microglia responses in human models of Alzheimer’s disease. Nat Commun. 2020;11(1):5370. doi:10.1038/s41467-020-19227-5
- Zheng H, Jia L, Liu CC, et al. TREM2 promotes microglial survival by activating Wnt/beta-Catenin pathway. J Neurosci. 2017;37(7):1772–1784. doi:10.1523/JNEUROSCI.2459-16.2017
- Ding X, Wang J, Huang M, et al. Loss of microglial SIRPalpha promotes synaptic pruning in preclinical models of neurodegeneration. Nat Commun. 2021;12(1):2030. doi:10.1038/s41467-021-22301-1
- Lehrman EK, Wilton DK, Litvina EY, et al. CD47 protects synapses from excess microglia-mediated pruning during development. Neuron. 2018;100(1):120–134. doi:10.1016/j.neuron.2018.09.017
- Wu C, Yang L, Youngblood H, Liu TC, Duan R. Microglial SIRPalpha deletion facilitates synapse loss in preclinical models of neurodegeneration. Neurosci Bull. 2022;38(2):232–234. doi:10.1007/s12264-021-00795-5
- Zhong L, Sheng X, Wang W, et al. TREM2 receptor protects against complement-mediated synaptic loss by binding to complement C1q during neurodegeneration. Immunity. 2023;56(8):1794–1808. doi:10.1016/j.immuni.2023.06.016
- Yin C, Ackermann S, Ma Z, et al. ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat Med. 2019;25(3):496–506. doi:10.1038/s41591-018-0336-8
- Zhang W, Xiao D, Mao Q, Xia H. Role of neuroinflammation in neurodegeneration development. Signal Transduct Target Ther. 2023;8(1):267. doi:10.1038/s41392-023-01486-5
- Ferri E, Rossi PD, Geraci A, Ciccone S, Cesari M, Arosio B. The sTREM2 concentrations in the blood: a marker of neurodegeneration? Front Mol Biosci. 2020;7:627931. doi:10.3389/fmolb.2020.627931
- Gu L, Shu H, Wang Y. Soluble TREM2 in body fluid in Alzheimer’s disease and Parkinson’s disease. Neurol Sci. 2023;44(8):2743–2751. doi:10.1007/s10072-023-06729-5
- Zhang X, Zhong X, Wang L, et al. Effects of soluble TREM2 on motor progression in Parkinson’s disease. Neurosci Lett. 2023;807:137277. doi:10.1016/j.neulet.2023.137277
- van der Ende EL, Morenas-Rodriguez E, Mcmillan C, et al. CSF sTREM2 is elevated in a subset in GRN-related frontotemporal dementia. Neurobiol Aging. 2021;103:151–158. doi:10.1016/j.neurobiolaging.2021.02.024
- Weber GE, Khrestian M, Tuason ED, et al. Peripheral sTREM2-Related inflammatory activity alterations in Early-Stage Alzheimer’s disease. J Immunol. 2022;208(10):2283–2299. doi:10.4049/jimmunol.2100771
- Rauchmann BS, Schneider-Axmann T, Alexopoulos P, Perneczky R. CSF soluble TREM2 as a measure of immune response along the Alzheimer’s disease continuum. Neurobiol Aging. 2019;74:182–190. doi:10.1016/j.neurobiolaging.2018.10.022
- Krasemann S, Madore C, Cialic R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47(3):566–581. doi:10.1016/j.immuni.2017.08.008
- Zhong L, Xu Y, Zhuo R, et al. Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model. Nat Commun. 2019;10(1):1365. doi:10.1038/s41467-019-09118-9
- Carosi JM, Sargeant TJ. Rapamycin and Alzheimer disease: a double-edged sword? Autophagy. 2019;15(8):1460–1462. doi:10.1080/15548627.2019.1615823
- Ising C, Venegas C, Zhang S, et al. NLRP3 inflammasome activation drives tau pathology. Nature. 2019;575(7784):669–673. doi:10.1038/s41586-019-1769-z
- van Olst L, Verhaege D, Franssen M, et al. Microglial activation arises after aggregation of phosphorylated-tau in a neuron-specific P301S tauopathy mouse model. Neurobiol Aging. 2020;89:89–98. doi:10.1016/j.neurobiolaging.2020.01.003
- Griciuc A, Serrano-Pozo A, Parrado AR, et al. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78(4):631–643. doi:10.1016/j.neuron.2013.04.014
- Karch CM, Jeng AT, Nowotny P, Cady J, Cruchaga C, Goate AM. Expression of novel Alzheimer’s disease risk genes in control and Alzheimer’s disease brains. PLoS One. 2012;7(11):e50976. doi:10.1371/journal.pone.0050976
- Nomura S, Tandon NN, Nakamura T, Cone J, Fukuhara S, Kambayashi J. High-shear-stress-induced activation of platelets and microparticles enhances expression of cell adhesion molecules in THP-1 and endothelial cells. Atherosclerosis. 2001;158(2):277–287. doi:10.1016/s0021-9150(01)00433-6
- Ramsborg CG, Papoutsakis ET. Global transcriptional analysis delineates the differential inflammatory response interleukin-15 elicits from cultured human T cells. Exp Hematol. 2007;35(3):454–464. doi:10.1016/j.exphem.2006.11.013
- Griciuc A, Federico AN, Natasan J, et al. Gene therapy for Alzheimer’s disease targeting CD33 reduces amyloid beta accumulation and neuroinflammation. Hum Mol Genet. 2020;29(17):2920–2935. doi:10.1093/hmg/ddaa179
- Jiang T, Yu JT, Hu N, Tan MS, Zhu XC, Tan L. CD33 in Alzheimer’s disease. Mol Neurobiol. 2014;49(1):529–535. doi:10.1007/s12035-013-8536-1
- Malik M, Simpson JF, Parikh I, et al. CD33 Alzheimer’s risk-altering polymorphism, CD33 expression, and exon 2 splicing. J Neurosci. 2013;33(33):13320–13325. doi:10.1523/JNEUROSCI.1224-13.2013
- Raj T, Ryan KJ, Replogle JM, et al. CD33: increased inclusion of exon 2 implicates the Ig V-set domain in Alzheimer’s disease susceptibility. Hum Mol Genet. 2014;23(10):2729–2736. doi:10.1093/hmg/ddt666
- Tortora F, Rendina A, Angiolillo A, et al. CD33 rs2455069 SNP: correlation with Alzheimer’s disease and hypothesis of functional role. Int J Mol Sci. 2022;23(7):3629. doi:10.3390/ijms23073629
- Eskandari-Sedighi G, Jung J, Macauley MS. CD33 isoforms in microglia and Alzheimer’s disease: friend and foe. Mol Aspects Med. 2023;90:101111. doi:10.1016/j.mam.2022.101111
- Malik M, Chiles JR, Xi HS, et al. Genetics of CD33 in Alzheimer’s disease and acute myeloid leukemia. Hum Mol Genet. 2015;24(12):3557–3570. doi:10.1093/hmg/ddv092
- Griciuc A, Patel S, Federico AN, et al. TREM2 acts downstream of CD33 in modulating microglial pathology in Alzheimer’s disease. Neuron. 2019;103(5):820–835. doi:10.1016/j.neuron.2019.06.010
- Salminen A, Kaarniranta K, Kauppinen A. Hypoxia/ischemia impairs CD33 (Siglec-3)/TREM2 signaling: potential role in Alzheimer’s pathogenesis. Neurochem Int. 2021;150(105186):105186. doi:10.1016/j.neuint.2021.105186
- Foltz IN, Karow M, Wasserman SM. Evolution and emergence of therapeutic monoclonal antibodies: what cardiologists need to know. Circulation. 2013;127(22):2222–2230. doi:10.1161/CIRCULATIONAHA.113.002033
- Assi R, Kantarjian H, Ravandi F, Daver N. Immune therapies in acute myeloid leukemia: a focus on monoclonal antibodies and immune checkpoint inhibitors. Curr Opin Hematol. 2018;25(2):136–145. doi:10.1097/MOH.0000000000000401
- Egan PC, Reagan JL. The return of gemtuzumab ozogamicin: a humanized anti-CD33 monoclonal antibody-drug conjugate for the treatment of newly diagnosed acute myeloid leukemia. Onco Targets Ther. 2018;11:8265–8272. doi:10.2147/OTT.S150807
- Jurcic JG. What happened to anti-CD33 therapy for acute myeloid leukemia? Curr Hematol Malig Rep. 2012;7(1):65–73. doi:10.1007/s11899-011-0103-0
- Lambert J, Pautas C, Terre C, et al. Gemtuzumab ozogamicin for de novo acute myeloid leukemia: final efficacy and safety updates from the open-label, Phase III ALFA-0701 trial. Haematologica. 2019;104(1):113–119. doi:10.3324/haematol.2018.188888
- Zhao L. CD33 in Alzheimer’s disease - biology, pathogenesis, and therapeutics: a Mini-Review. Gerontology. 2019;65(4):323–331. doi:10.1159/000492596
- Feng YS, Tan ZX, Wu LY, Dong F, Zhang F. The involvement of NLRP3 inflammasome in the treatment of Alzheimer’s disease. Ageing Res Rev. 2020;64(101192). doi:10.1016/j.arr.2020.101192
- Jha D, Bakker E, Kumar R. Mechanistic and therapeutic role of NLRP3 inflammasome in the pathogenesis of Alzheimer’s disease. J Neurochem. 2023. doi:10.1111/jnc.15788
- Halle A, Hornung V, Petzold GC, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9(8):857–865. doi:10.1038/ni.1636
- Bai H, Zhang Q. Activation of NLRP3 inflammasome and onset of Alzheimer’s disease. Front Immunol. 2021;12(701282). doi:10.3389/fimmu.2021.701282
- Heneka MT, Kummer MP, Stutz A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493(7434):674–678. doi:10.1038/nature11729
- Cai Y, Chai Y, Fu Y, et al. Salidroside ameliorates Alzheimer’s disease by targeting NLRP3 Inflammasome-Mediated pyroptosis. Front Aging Neurosci. 2021;13:809433. doi:10.3389/fnagi.2021.809433
- Chiu YJ, Lin CH, Lee MC, et al. Formulated Chinese medicine Shaoyao Gancao Tang reduces NLRP1 and NLRP3 in Alzheimer’s disease cell and mouse models for neuroprotection and cognitive improvement. Aging. 2021;13(11):15620–15637. doi:10.18632/aging.203125
- Xu Z, Zhou X, Hong X, et al. Essential oil of Acorus tatarinowii Schott inhibits neuroinflammation by suppressing NLRP3 inflammasome activation in 3 x Tg-AD transgenic mice. Phytomedicine. 2023;112:154695. doi:10.1016/j.phymed.2023.154695
- Ben-Menachem-Zidon O, Ben-Menahem Y, Ben-Hur T, Yirmiya R. Intra-hippocampal transplantation of neural precursor cells with transgenic over-expression of IL-1 receptor antagonist rescues memory and neurogenesis impairments in an Alzheimer’s disease model. Neuropsychopharmacol. 2014;39(2):401–414. doi:10.1038/npp.2013.208
- Fernandez PL, Britton GB, Rao KS. Potential immunotargets for Alzheimer’s disease treatment strategies. J Alzheimers dis. 2013;33(2):297–312. doi:10.3233/JAD-2012-121222
- Craft JM, Watterson DM, Frautschy SA, Van Eldik LJ. Aminopyridazines inhibit beta-amyloid-induced glial activation and neuronal damage in vivo. Neurobiol Aging. 2004;25(10):1283–1292. doi:10.1016/j.neurobiolaging.2004.01.006
- Craft JM, Van Eldik LJ, Zasadzki M, Hu W, Watterson DM. Aminopyridazines attenuate hippocampus-dependent behavioral deficits induced by human beta-amyloid in a murine model of neuroinflammation. J Mol Neurosci. 2004;24(1):115–122. doi:10.1385/JMN:24:1:115
- Hu W, Ralay RH, Craft JM, Van Eldik LJ, Watterson DM. Validation of the neuroinflammation cycle as a drug discovery target using integrative chemical biology and lead compound development with an Alzheimer’s disease-related mouse model. Curr Alzheimer Res. 2005;2(2):197–205. doi:10.2174/1567205053585828
- Mirzoeva S, Sawkar A, Zasadzki M, et al. Discovery of a 3-amino-6-phenyl-pyridazine derivative as a new synthetic antineuroinflammatory compound. J Med Chem. 2002;45(3):563–566. doi:10.1021/jm015573g
- Vandenabeele P, Fiers W. Is amyloidogenesis during Alzheimer’s disease due to an IL-1-/IL-6-mediated ‘acute phase response’ in the brain? Immunol Today. 1991;12(7):217–219. doi:10.1016/0167-5699(91)90032-O
- Mrak RE, Griffin WS. Interleukin-1, neuroinflammation, and Alzheimer’s disease. Neurobiol Aging. 2001;22(6):903–908. doi:10.1016/s0197-4580(01)00287-1
- Huang Z, Xie L, Li H, et al. Insight into interleukin-37: the potential therapeutic target in allergic diseases. Cytokine Growth Factor Rev. 2019;49:32–41. doi:10.1016/j.cytogfr.2019.10.003
- Terrando N, Rei FA, Vizcaychipi M, et al. The impact of IL-1 modulation on the development of lipopolysaccharide-induced cognitive dysfunction. Crit Care. 2010;14(3):R88. doi:10.1186/cc9019
- Craft JM, Watterson DM, Hirsch E, Van Eldik LJ. Interleukin 1 receptor antagonist knockout mice show enhanced microglial activation and neuronal damage induced by intracerebroventricular infusion of human beta-amyloid. J Neuroinflammation. 2005;2:15. doi:10.1186/1742-2094-2-15
- Huang ZB, Sheng GQ. Interleukin-1beta with learning and memory. Neurosci Bull. 2010;26(6):455–468. doi:10.1007/s12264-010-6023-5
- Lynch MA. Neuroinflammatory changes negatively impact on LTP: a focus on IL-1beta. Brain Res. 2015;1621:197–204. doi:10.1016/j.brainres.2014.08.040
- Kitazawa M, Cheng D, Tsukamoto MR, et al. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer’s disease model. J Immunol. 2011;187(12):6539–6549. doi:10.4049/jimmunol.1100620
- Sutinen EM, Pirttila T, Anderson G, Salminen A, Ojala JO. Pro-inflammatory interleukin-18 increases Alzheimer’s disease-associated amyloid-beta production in human neuron-like cells. J Neuroinflammation. 2012;9:199. doi:10.1186/1742-2094-9-199
- Ojala J, Alafuzoff I, Herukka SK, van Groen T, Tanila H, Pirttila T. Expression of interleukin-18 is increased in the brains of Alzheimer’s disease patients. Neurobiol Aging. 2009;30(2):198–209. doi:10.1016/j.neurobiolaging.2007.06.006
- Ojala JO, Sutinen EM, Salminen A, Pirttila T. Interleukin-18 increases expression of kinases involved in tau phosphorylation in SH-SY5Y neuroblastoma cells. J Neuroimmunol. 2008;205(1–2):86–93. doi:10.1016/j.jneuroim.2008.09.012
- Zhang J, Song T, Liang H, Lian J, Zhang G, Gong H. Interleukin-18 −137 G/C and −607 C/A polymorphisms and Alzheimer’s disease risk: a meta-analysis. Neurol Sci. 2016;37(6):921–927. doi:10.1007/s10072-016-2516-y
- Boyd RJ, Avramopoulos D, Jantzie LL, Mccallion AS. Neuroinflammation represents a common theme amongst genetic and environmental risk factors for Alzheimer and Parkinson diseases. J Neuroinflammation. 2022;19(1):223. doi:10.1186/s12974-022-02584-x
- Huang LK, Chao SP, Hu CJ. Clinical trials of new drugs for Alzheimer disease. J Biomed Sci. 2020;27(1):18. doi:10.1186/s12929-019-0609-7
- Huang LK, Kuan YC, Lin HW, Hu CJ. Clinical trials of new drugs for Alzheimer disease: a 2020–2023 update. J Biomed Sci. 2023;30(1):83. doi:10.1186/s12929-023-00976-6
- Cui W, Sun C, Ma Y, Wang S, Wang X, Zhang Y. Inhibition of TLR4 Induces M2 Microglial Polarization and Provides Neuroprotection via the NLRP3 Inflammasome in Alzheimer’s Disease. Front Neurosci. 2020;14:444. doi:10.3389/fnins.2020.00444
- Gold M, Alderton C, Zvartau-Hind M, et al. Rosiglitazone monotherapy in mild-to-moderate Alzheimer’s disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cognit Disord. 2010;30(2):131–146. doi:10.1159/000318845
- Swardfager W, Lanctôt K, Rothenburg L, Wong A, Cappell J, Herrmann N. A meta-analysis of cytokines in Alzheimer’s disease. Biol Psychiatry. 2010;68(10):930–941. doi:10.1016/j.biopsych.2010.06.012
- Lai KSP, Liu CS, Rau A, et al. Peripheral inflammatory markers in Alzheimer’s disease: a systematic review and meta-analysis of 175 studies. J Neurol Neurosurg Psychiatry. 2017;88(10):876–882. doi:10.1136/jnnp-2017-316201
- Lananna BV, McKee CA, King MW, et al. S. Chi3l1/YKL-40 is controlled by the astrocyte circadian clock and regulates neuroinflammation and Alzheimer’s disease pathogenesis. Sci Transl Med. 2020;12(574):eaax3519. doi:10.1126/scitranslmed.aax3519
- Alcolea D, Martínez-Lage P, Sánchez-Juan P, et al. Amyloid precursor protein metabolism and inflammation markers in preclinical Alzheimer disease. Neurology. 2015;85(7):626–633. doi:10.1212/WNL.0000000000001859
- Stefaniak J, O’Brien J. Imaging of neuroinflammation in dementia: a review. J Neurol Neurosurg Psychiatry. 2016;87(1):21–28. doi:10.1136/jnnp-2015-311336
- Parbo P, Ismail R, Hansen KV, et al. Brain inflammation accompanies amyloid in the majority of mild cognitive impairment cases due to Alzheimer’s disease. Brain. 2017;140(7):2002–2011. doi:10.1093/brain/awx120
- Chen H, Guo Z, Gao X, Dai X. Progress in investigating the role of microglia in Alzheimer’s disease. Chinese Bulletin of Life Sic. 2022;34(07):830–837. doi:10.13376/j.cbls/2022091