 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Background
H1-antihistamines are recommended as the first-line symptomatic treatment of allergic rhinitis. The objective of this study was to evaluate the effects of rupatadine (RUP) versus desloratadine (DES) in subjects with seasonal allergic rhinitis (SAR).
Method
To assess the efficacy and safety of RUP in SAR in comparison with placebo (PL) and DES. A randomized, double-blind, multicenter, international, and PL-controlled study was carried out. The main selection criteria included SAR patients over 12 years old with a positive prick test to a relevant seasonal allergen for the geographic area. Symptomatic patients at screening with a nasal symptom sum score of ≥6 points (nasal discharge, nasal obstruction, sneezing, and nasal pruritus), a non-nasal score of ≥3 points (ocular pruritus, ocular redness, and tearing eyes), and a rhinorrhea score of ≥2 points with laboratory test results and electrocardiography within acceptable limits were included in the study. Change from baseline in the total symptom-score (T7SS) over the 4-week treatment period (reflective evaluation) was considered the primary efficacy variable. Secondary efficacy measures included total nasal symptom score (T4NSS) and conjunctival symptom score (T3NNSS), both of which are reflective and instantaneous evaluations. Furthermore questions related to quality of life (eg, sleep disturbances or impairment of daily activities) have also been evaluated. Safety was assessed according to adverse events reported, as well as laboratory and electrocardiography controls.
Results
A total of 379 patients were randomized, of which 356 were included and allocated to PL (n = 122), RUP (n = 117), or DES (n = 117). Mean change of T7SS over the 4-week treatment period was significantly reduced in the RUP (–46.1%, P = 0.03) and DES (–48.9%, P = 0.01) groups, compared with PL. Similarly, RUP and DES were comparable and significantly superior to PL for all secondary endpoints, including nasal and conjunctival symptoms and patients’ and investigator’s overall clinical opinions. Symptom score evaluation (both reflective and instantaneous evaluations) throughout the treatment period showed a progressive and maintained significant improvement with both treatments at day 7 (P = 0.01), day 14 (P = 0.007), and day 21 (P = 0.01) in comparison with PL. Adverse events were scarce and were similar in both treatment groups. Electrocardiography (QTc) and lab test results did not show any relevant findings
Conclusion
RUP is a very good choice for SAR due to its contribution to the improvement of nasal (including obstruction) and non-nasal symptoms to a similar degree as DES.
Introduction
Allergic rhinitis (AR) is an increasingly prevalent disease in the majority of developed countries, affecting 23%–30% of the general population in Europe.Citation1–Citation3 In some countries it is now approaching epidemic proportions and is becoming a significant public health concern.Citation4 AR affects patients of all ages and ethnic groups, causes major illness like predisposal to asthma and disability worldwide, and affects an individual’s social life, sleep, school, and work.Citation5 AR still remains undermanaged and an analysis of both control and clearly defined severity phenotypes may help contribute to better therapy approaches.
Second generation oral H1-antihistamines have been the traditional first-line therapy in the treatment of ARs against symptoms mediated by histamine (rhinorrhea, sneezing, nasal itching, and eye symptoms), but they have been less effective with nasal congestion.Citation6 The primary goal of treating patients with AR is to provide symptomatic relief. At the present time, the market is flooded with a lot of similar H1 compounds, and physicians are overwhelmed with such a great amount of promotional literature that has scarce comparative data among the different therapeutic agents.
Rupatadine is a powerful once-daily nonsedating H1 antihistamine. It has also been found to inhibit platelet-activating factor (PAF) through its interactions with specific receptors.Citation7,Citation8 PAF and histamine are known to complement each other in vivo; histamine is a mediator of early response, being released from preformed reservoirs in mast cells, whereas PAF is mainly synthesized de novo in response to the allergic stimulus.Citation9 Previous studies have proven that PAF is also an important mediator of AR. PAF causes vasodilatation and an increase in vascular permeability that may contribute to the appearance of rhinorrhea and nasal congestion.Citation10 Rupatadine is well known as a dual blocker of histamine H1 and PAF-receptors, by means of a variety of experimental and clinical studies which provide scientific evidence that this can be an effective and well tolerated treatment for AR and urticaria.Citation11,Citation12
The objective of the current study was to compare the efficacy and safety of rupatadine 10 mg versus a very well known second generation H1 antihistamine such as desloratadine 5 mg, and to also compare the efficacy and safety of both drugs versus placebo, all administered once daily, in the treatment of SAR over a 4-week period. This will provide the first clinical evidence of a direct comparison between both types of H1-receptor antagonists.
Methods
Design of the study and treatments assessed
This was a randomized, double-blind, parallel-group, placebo-controlled, comparative study of desloratadine 5 mg and rupatadine 10 mg. All three treatments (two active and the placebo) were administered orally in identical timetables each morning within 1 hour after waking up. A total of 26 centers in France (n = 5), Germany (n = 4), Poland (n = 6), Romania (n = 7), and Spain (n = 4) participated in the trial. The trial was conducted in compliance with local ethical committees and good clinical practice guidelines, as well as with local clinical trial regulations. All patients gave their written informed consent before being included in the study.
Inclusion and exclusion criteria
Patients were diagnosed with SAR. The main criteria for inclusion were: male or female aged older than 12 years; having a documented history of SAR at least 2 years before the screening date with a positive prick test performed on the same day or within 1 year before the screening visit; being clinically symptomatic at screening with a nasal symptom sum score ≥ 6 points, non-nasal score ≥ 3 points, and rhinorrhea score ≥ 2 points; and with results from laboratory tests and electrocardiography within acceptable limits. Furthermore, the QTc interval values (milliseconds) after Bazzet’s correction had to be normal (not prolonged). The values considered to be normal were ,430 milliseconds for males and ,450 milliseconds for females.
Patients were selected for randomization based on reflective self-assessments on a screening diary card completed within the last 3 consecutive days, where they were required to present a nasal discharge sum score ≥ 12, a total nasal sum score ≥ 36, and a total non-nasal sum score ≥ 18. Additionally if a patient was on a regularly scheduled immunotherapy or who had mild asthma symptoms that were being treated with inhaled bronchodilators, he or she was allowed to be included in the study. Finally, women of childbearing age had to have a negative pregnancy test and use contraceptive measures.
Patients suffering from non-AR (eg, vasomotor, infectious, or drug-induced rhinitis) or with a negative prick test were not included. Patients being treated with nasal decongestants in the previous 24 hours, oral antihistamines, or disodium cromoglicate (in the previous week), ketotifen (in the previous month), topical antihistamines (in the previous 48 hours), systemic or topical treatment with corticosteroids (except for topical hydrocortisone < 1%), immunosuppressants, or any investigational drug within 2 weeks prior to inclusion, were also excluded. Other relevant exclusion criteria included abnormal laboratory values of clinical significance, certain conditions that may interfere with response to treatment such as: mild asthma treated with inhaled bronchodilators or inhaled corticosteroids > 800 mcg/day of budesonide or beclomethasone; with >.500 mcg/day of fluticasone; obstructive nasal polyps; or hypersensitivity to compounds structurally related to the study drug.
Evaluation of efficacy on symptoms score
At the screening visit, patients were instructed by the investigator to self-evaluate their scores via the daily diary twice each day, first in the morning just before taking medication and approximately 12 hours later, in the evening. Both evaluations should be reflective, based on the patient’s evaluation of symptom severity in the previous 12 hours, and based on the score at the time of recording, which took place at the same time as the patient’s evaluation.
Each patient received a diary card for the daily recording of symptoms at the start of the treatment. Severity scores for seven (T7SS) individual AR signs/symptoms; nasal (nasal discharge, nasal obstruction, sneezing, and nasal pruritus) and non-nasal symptoms (ocular pruritus, ocular redness, and tearing eyes) were recorded on the diary card and scored numerically on a scale from 0–3, where 0 indicates no symptoms present, 1 indicates mild symptoms (occasionally present, but not troublesome), 2 indicates moderate symptoms (frequently present and annoying), and 3 indicates severe symptoms (continuously present and interfering with work or sleep).
The investigators examined the patients’ diary cards at each follow-up visit (days 14 ± 3 and 28 ± 3) to check treatment compliance and to give any advice as necessary.
Change from baseline in the total patient symptom score (T7SS) over the 4-week treatment period (reflective evaluation) was considered the primary efficacy variable. Furthermore, secondary variables were also calculated: the change from baseline in the T7SS over the 4-week treatment period (instantaneous evaluation), change from baseline in the total nasal symptom score (T4NSS) and non-nasal symptom score (T3NNSS), which are both reflective and instantaneous evaluations.
Subject’s and investigator’s evaluation of therapeutic response
The subject’s and investigator’s evaluation of therapeutic response to treatment at 2 and 4 weeks was also assessed. In these two follow-up visits, the patients’ and the physician’s global evaluation of efficacy were scored numerically. Scores of symptom severity from visit 0 were measured on a scale of 0 (worsened), 1 (no change), 2 (slight improvement), 3 (good improvement), and 4 (excellent improvement).
Evaluation of treatment compliance
Treatment compliance was assessed by means of drug accountability at days 14 and 28 using the following formula:
and was considered as one of the criteria for major protocol deviations. The definition of incorrectly administered treatment or noncompliance included patients who were missing four or more tablets in each of the treatment boxes (assessed on visit two and the last visit), or who experienced overdose (more than four tablets per day).
Evaluation assessment of sleep disturbances and impairment of daily activities
The investigators assessed at each visit and at the end of treatment the impact on sleep disturbances and impairment of daily activities using a four-point scale ranging from 0–3, with 0 = none, 1 = mild, 2 = moderate, and 3 = severe.
Evaluation of safety
Treatment safety and tolerability were evaluated according to the incidence and type of adverse events spontaneously reported in the patients’ diaries, or were reported as an answer to the investigator’s question, “Have you noticed any discomfort during these days?” that was asked at each visit.
Laboratory safety tests (complete blood count and standard serum chemistry profile) and physical examinations (all of which were performed during the study as well as at the end of the study period) were considered. All adverse events were coded using the World Health Organization Adverse Reactions terminology dictionary, and grouped by treatment.Citation13
Statistical analysis
The study was designed to enroll 366 patients (sample size of 122 per treatment group and three treatment groups). This figure was chosen to detect, with 80% power and at a 5% significance level, a difference ≥ 1.8 units in the primary efficacy variable, assuming a standard deviation of 4.45 and a loss rate of 20%.Citation14
Analysis of variance (ANOVA by country and treatment) or analysis of covariance (including treatment, country, and baseline values as covariances) were used to compare treatment groups for the quantitative primary and secondary outcomes. In the event that significant results were observed, subsequent pairwise comparisons using a Bonferroni adjustment were made between the treatment groups. For quantitative (efficacy and safety) variables, mean, median, standard deviation, maximum, and minimum values were calculated.
Qualitative variables were expressed as relative frequencies. The Chi-square test was used for the qualitative variables, and Fisher’s exact test was used if the applicability conditions were not present. The Mantel–Haenszel Chi-square test was performed in case both variables were on an ordinal scale.
Analysis of both efficacy and safety was based on the intention to treat population (ITT), including all patients who were randomized and received at least one dose of the study medication. The adverse events were coded using the Medical Dictionary for Regulatory Activities (MedDRA® v 12.1, MedDRA Maintenance and Support Services Organization, Chantilly, Virginia), and the incidence of adverse events was compared between treatment groups using the Chi-square test.
All statistical analyses were two-tailed, with a significance level set at P < 0.05. Statistical analyses were performed using the SAS® statistical software (SAS Institute Inc, Cary, NC, USA).
Results
Study population and baseline characteristics
From April 2005 to August 2005, 450 patients were selected in 26 centers in Europe, distributed in the following countries: France (n = 5), Germany (n = 4), Poland (n = 6), Romania (n = 7), and Spain (n = 4). Out of those 450 patients, 359 were randomized into three different groups: placebo (n = 122), desloratadine 5 mg (n = 119), and rupatadine 10 mg (n = 118). The global disposition of patients during the study is shown in .
Figure 1 Global disposition of patients during the study.
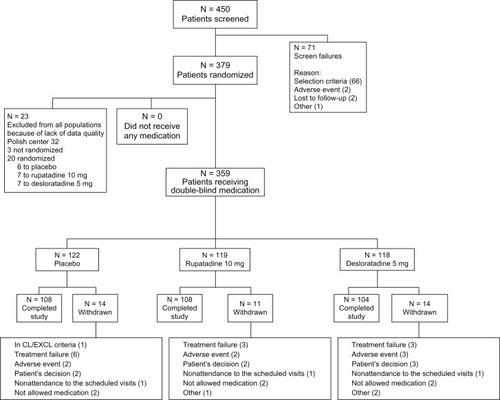
The ITT included all randomized subjects who received any study drug, and for whom one postbaseline efficacy value was present, regardless of the degree of adherence to the protocol. Finally, ITT analysis for efficacy was considered in 356 patients (122, placebo; 117 rupatadine; 117 desloratadine) due to 3 cases were detected on diary cards have not included on diary cards efficacy measurements.
At baseline, all groups were similar and showed no significant differences in the demographic data and clinical characteristics of the patients ().
Table 1 Demographic and clinical characteristics at baseline in the ITT population
The mean treatment compliance percentage was 98.5% for placebo, 97.1% for rupatadine 10 mg, and 96.9% for desloratadine 5 mg, with no statistically significant difference between treatment groups (ANOVA; P = 0.405). No patients took any rescue medication during the treatment period.
Efficacy measures
represents the ITT analysis for the end-point indexes at baseline and after 4 weeks of treatment, considering the absolute values and the percentage of reduction from baseline.
Table 2 Effect of treatments on total symptom (T7SS), nasal symptom (T4NSS), and non-nasal symptom (T3NNSS) scores in the ITT population at 4 weeks
Total patient symptom score (T7SS) over the 4-week treatment period (reflective evaluation), confirmed the globally different effectiveness among the three groups (analysis of covariance; P = 0.002) (). These differences were distributed between rupatadine 10 mg (ANOVA; P = 0.03) and desloratadine 5 mg (ANOVA; P = 0.01), both of which were shown to be more effective than placebo in reducing the baseline symptoms. This means that there were symptom reductions of 46.1% and 48.9% with rupatadine and desloratadine, respectively. There were not statistically significant differences between rupatadine and desloratadine in reducing the baseline symptoms.
Figure 2 Mean change from baseline of reflective and instantaneous values for T7SS at 4 weeks.
Abbreviation: T7SS, total seven symptoms score.
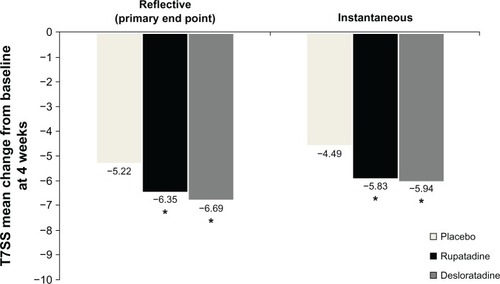
In the same way, active groups showed significant differences in the effectiveness (reflective evaluation) in reducing the nasal symptoms (T4NSS) from baseline in comparison to placebo (P = 0.03). When non-nasal symptoms (T3NNSS) were also considered, rupatadine and desloratadine were more effective than placebo (P = 0.024, P = 0.005, respectively). No significant changes were detected between the active groups ().
Rupatadine and desloratadine were more effective than placebo in reducing baseline nasal discharge (P = 0.03, P = 0.02, respectively), sneezing (both values, P = 0.01), nasal pruritus (P = 0.05, P = 0.003, respectively), and ocular pruritus (P = 0.002, P < 0.001, respectively) (). Rupatadine 10 mg did not show different effectiveness compared to desloratadine 5 mg in reducing any of the baseline symptoms.
Figure 3 Mean change from baseline of reflective and instantaneous values of daily individual symptoms score at 4 weeks.
Abbreviations: N, nasal; O, ocular.
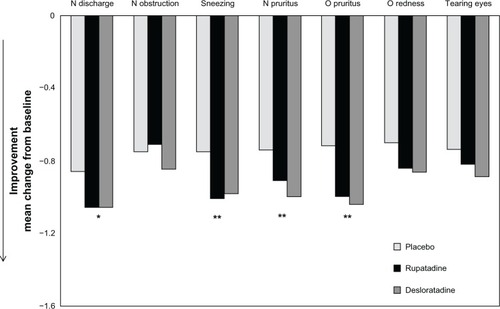
The evaluation of symptom scores (reflective evaluations) showed a progressive and significant improvement with both treatments in comparison to placebo during the treatment period at day 7 (P = 0.01), 14 (P = 0.007), or day 21 (P = 0.01). Rupatadine 10 mg did not show different effectiveness compared to desloratadine 5 mg. Similar results have been obtained when analyzing instantaneous assessments of symptoms of the two active drugs.
Regarding the onset of action, which is defined as the first day where there were differences among treatments, rupatadine showed significant differences when compared to placebo (P < 0.05). The onset of action of desloratadine appeared 1 day later.
Regarding the investigators’ clinical impression, both rupatadine and desloratadine presented higher investigator therapeutic response assessments compared to placebo at the last visit (ANOVA; P = 0.038, P = 0.007, respectively). In the same way, regarding the patients’ assessments, both rupatadine and desloratadine showed a greater patient therapeutic response compared to placebo at the last visit (ANOVA; P = 0.018, P < 0.001, respectively). In both ANOVA analyses, there was an interaction between treatment and country across investigators’ clinical impression variable.
Finally, regarding sleep disturbances, some differences among treatment groups have been confirmed at 2 and 4 weeks. Regarding the impairment of daily activities, rupatadine showed a lower impact of impairment of daily activities compared to placebo at 4 weeks (ANOVA; P = 0.027). In the same way, desloratadine provided no impact in daily activities at the first week (P = 0.015) and at 4 weeks (P = 0.04).
Safety
The adverse events incidences were 36.1% for patients taking placebo, 39.5% for rupatadine 10 mg, and 37.3% for desloratadine 5 mg. There were no differences observed among treatments (Chi-square; P = 0.857). The most common related AEs are categorized by incidence ≥2% in patients were reported in each treatment group are showed on . The related adverse events were 9.8% for those in the placebo group, 13.4% for those in the rupatadine 10 mg group, and 16.1% for those in the desloratadine 5 mg group. There were no differences observed among treatments (chi square; P = 0.352).
Table 3 Incidence of adverse events is ≥2% as reported during the study by treatment group
No differences were detected between placebo and rupatadine regarding the percentage of patients who experienced an increase of the QTcB and/or QTcF parameters at the last visit from baseline. When we analized the number or percentage of patients whose increased (msec) with respect QTcB and QTcF baseline values, no differences were detected among treatments categorized below 30 msec increase, between 30 and 60 msec increase and over 60 msec increase.
Discussion
This study has confirmed that rupatadine 10 mg was more effective than placebo in reducing the baseline symptoms in seasonal allergic rhinoconjunctivitis during a period of 4 weeks. Furthermore, no differences were found between rupatadine 10 mg compared to desloratadine 5 mg. Also desloratadine 5 mg was superior to placebo. The efficacy result (primary endpoint) was supported with different sensitivity analyses, which explored the implications of considering different factors like variability from baseline values, geographic location, or considering the different imputation methods to assess potential missing values that occur very frequently in these allergy studies.
Using the reflective assessments as a primary endpoint, rupatadine 10 mg reduced 46% of the baseline symptoms; compared to desloratadine 5 mg which reduced 49% of baseline symptoms, and placebo which reduced the baseline symptoms in 37% of patients. Similar results were obtained in the instantaneous evaluation of patients. These percentages of symptom reductions associated with placebo and desloratadine are very close to those obtained in a recent clinical study using a similar methodology and conducted in similar participating countries among SAR patients in a shorter period of evaluation.Citation15 It is also remarkable that the improvement in symptoms from baseline were observed among the placebo group of >35% across all symptoms, including ocular symptoms, considering that the baseline values of this group were similar to those of the active treatments. This observation is not unusual since it has been previously described with other anti-H1 compounds in clinical trials.Citation16,Citation17 However, in spite of the remarkable placebo effect, the difference between the placebo and active groups was significant, as soon as in the first 5 days and, more importantly, it remained significant both at 2 and 4 weeks for the primary and in many of the relevant secondary endpoints.
Both rupatadine and desloratadine improved both investigator and patients’ therapeutic response assessments compared to placebo at 4 weeks. Furthermore, there were no differences among the treatment groups in the assessment of sleep disturbances. The use of rupatadine 10 mg presented with a lower impairment of daily activities than placebo at the final visit, with no differences noted when compared to desloratadine 5 mg.
The close similarity between rupatadine and desloratadine had been expected in relation to the findings in the previous studies reported with SAR patients.Citation18 The findings of this study are in accordance with previous meta-analysis with desloratadine in comparison with placebo, involving several controlled clinical trials with higher degrees of variability.Citation19 Nevertheless, direct comparisons involving large samples are relatively infrequent in the literature,Citation20 and overall, there were no results significantly favoring levocetirizine or fexofenadine over desloratadine in terms of their effects on AR symptoms.
Despite the actual preference of Allergic Rhinitis and its Impact on Asthma classification, no previous studies with desloratadine or rupatadine were released before 2006 in patients with intermittent or persistent AR that could determine the efficacy based on the duration of the severity of symptoms and their impact on quality of life.Citation21–Citation23 When this study was designed, the AR patients were involved accordingly with the traditional classification of SAR given that the sample size being estimated was only being based on previous studies with SAR patients reported with desloratadine and rupatadine.Citation24–Citation27 Nevertheless, desloratadine, rupatadine, and levocetirizine are the only anti-H1 compounds that have been shown to be effective and safe under this Allergic Rhinitis and its Impact on Asthma classification.Citation28
From the point of view of safety, it can be concluded that the overall incidence of related adverse events with rupatadine was similar to the incidence of adverse events in patients treated with placebo and desloratadine. The electrocardiography analysis of the QTc parameters conducted in the study yielding findings that were similar between both anti-H1 compounds, and these were consistent with those findings observed in studies examining rupatadine in the evaluation of cardiac safety in humans following the most recent recommendations on this matter.Citation29,Citation30
In the present study, which was conducted in adolescents and adults, we have demonstrated that the daily administration of rupatadine is significantly more effective than placebo in relieving the symptoms of SAR throughout the 4 weeks of treatment. Given that rupatadine significantly improves the nasal and ocular symptoms of AR suggests that this agent may have significant advantages in addressing a wide range of rhinoconjunctivitis symptoms.
Study participants
France: Francois Wessel, Montserrat Angell-Perello, Laurent Fouquert, Sylvie Huet Francoise Sanquer. Germany: Karl Friedrich Lukat, Norbet Pasch, Ulrich Botzen, Manfred Wener, Lange Bernd. Poland: Marek L Kowalski, Edayta Kominek. Romania: Ioana Agache, Luminita Agachi, Adriana Iacomi. Spain: Alfonso del-Cuvillo, Daniel Muñoz, Pilar Rivas, Albert Roger.
Acknowledgments
The authors thank J Uriach y Compañía (Barcelona) for financial support for this study. We would like to thank Teodoro Sanchez for helping with the English editing of the paper.
Disclosure
The authors report no conflicts of interest in this work. Dr Iñaki Izquierdo is an employee of J Uriach y Compañía, SA, Catalonia, Spain. None of the other authors have any conflicts of interest.
References
- BauchauVDurhamSRPrevalence and rate of diagnosis of allergic rhinitis in EuropeEur Respir J200424575876415516669
- BachertCvan CauwenbergePOlbrechtJvan SchoorJPrevalence, classification and perception of allergic and nonallergic rhinitis in BelgiumAllergy200661669369816677237
- BousquetJKhaltaevNCruzAAWorld Health OrganizationGA(2)LEN, AllerGen. Allergic Rhinitis and its Impact on Asthma (ARIA) 2008 update (in collaboration with the World Health Organization, GA(2)LEN and AllerGen)Allergy200863Suppl 86816018331513
- SchatzMA survey of the burden of allergic rhinitis in the USAAllergy200762Suppl 8591617927673
- MullolJA survey of the burden of allergic rhinitis in SpainJ Invest Allergol Clin Immunol20091912734
- HoreIGeorgalasCScaddingGOral antihistamines for the symptom of nasal obstruction in persistent allergic rhinits – a systematic review of randomized controlled trialsClin Exp Allergy200535220721215725193
- MerlosMGiralMBalsaDRupatadine, a new potent, orally active dual antagonist of histamine and platelet-activating factor (PAF)J Pharmacol Exp Ther199728011141218996188
- QueraltMBrazisPMerlosMPuigdemontAInhibitory effects of rupatadine on mast cell histamine release and skin wheal development induced by Ascaris suum in hypersensitive dogsDrug Dev Res1998444955
- PiwinskiJJWongJKGreenMJDual antagonists of platelet activating factor and histamine. Identification of structural requirements for dual activity of N-Acyl-4-(5,6-dihydro-11H-benzo[5,6] cyclohepta-[1.2-b]pyridin-11-ylidene)piperidinesJ Med Chem19913414574611671420
- AlfaroVRole of histamine and platelet-activating factor in allergic rhinitsJ Phisiol Biochem2004602101111
- MullolJBousquetJBachertCRupatadine in allergic rhinitis and chronic urticariaAllergy200863 Suppl 8752818339040
- KeamSJPloskerGLRupatadine: a review of its use in the management of allergic disordersDrug2007673457474
- European medecines AgencyICH Topic E 6 (R1) Guideline for Good Clinical Practice Available from: http://www.emea.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002874.pdfAccessed 3 Jan 2013
- SalmunLMLorberR24-hour efficacy of once-daily desloratadine therapy in patients with seasonal allergic rhinitis [ISRCTN32042139]BMC Fam Pract200231412162793
- BachertCKunaPSanquerFBilastine International Working GroupComparison of the efficacy and safety of bilastine 20 mg vs desloratadine 5 mg in seasonal allergic rhinitis patientsAllerg y2009641158165
- SchenkelEJEffect of desloratadine on the control of morning symtoms in patients with seasonal and perennial allergic rhinitisAllergy Asthma Proc200627646547217176780
- Van CauwenbergePJuniperEFComparison of the efficacy, safety and quality of life provided by fexofenadine hydrochloride 120 mg, loratadine 10 mg and placebo administered once daily for the treatment of seasonal allergic rhinitisClin Exp Allergy200030689189910848909
- GehaRSMeltzerEODesloratadine: A new, nonsedating, oral antihistamineJ Allergy Clin Immunol2001107475176211295678
- CanonicaG WTarantiniFCompalatiEPenagosMEfficacy of desloratadine in the treatment of allergic rhinitis: a meta-analysis of randomized, double-blind, controlled trialsAllergy200762435936617362245
- FumagalliFBaiardiniIPasqualiMAntihistamines: do they work? Further well-controlled trials involving larger samples are neededAllergy200459 Suppl 78747715245363
- BousquetJBachertCCanonicaGWACCEPT-1 study groupEfficacy of desloratadine in intermittent allergic rhinitis: a GA(2)LEN studyAllerg y2009641015161523
- BachertCvan CauwenbergePDesloratadine treatment for intermittent and persistent allergic rhinitis: a reviewClin Ther20072991795180218035184
- FantinSMasperoJBisbalCInternational Rupatadine study groupA 12-week placebo-controlled study of rupatadine 10 mg once daily compared with cetirizine 10 mg once daily, in the treatment of persistent allergic rhinitisAllergy200863792493118588560
- KeithPKLuciukGEffectiveness of desloratadine 5 mg once daily in patients with symptoms of seasonal allergic rhinitis: results of a Canadian multicenter, open-label trialClin Ther200729341942617577463
- BachertCVirchowCJJrPlenkerADesloratadine in the treatment of seasonal allergic rhinitis: results of a large observational studyClin Drug Investig2002224352
- GuadañoEMSerra-BatllesJMeseguerJRupatadine Study GroupRupatadine 10 mg and ebastine 10 mg in seasonal allergic rhinitis: a comparison studyAllergy200459776677115180765
- Martinez-CóceraCDe MolinaMMartí-GuadañoESpanish Rupatadine Rhinitis Study GroupRupatadine 10 mg and cetirizine 10 mg in seasonal allergic rhinitis: a randomised, double-blind parallel studyJ Investig Allergol Clin Immunol20051512229
- MullolJPositioning of antihistamines in the Allergic Rhinitis and its Impact on Asthma (ARIA) guidelinesClinical and Experimental Allergy Reviews20121211726
- DonadoEIzquierdoIPérezINo cardiac effects of therapeutic and supratherapeutic doses of rupatadine: results from a ‘thorough QT/ QTc study’ performed according to ICH guidelinesBr J Clin Pharmacol201069440141020406224
- European Medicines AgencyICH Topic E 14. The Clinical Evaluation of QT/Qtc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic DrugsLondon, UKICH Steering Committee2005 Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientifc_guideline/2009/09/WC500002879.pdfAccessed Nov 2012