
Abstract
Hereditary angioedema (HAE) is a rare, chronic, and debilitating genetic disorder characterized by recurrent and unpredictable swelling episodes that primarily affect the subcutaneous and/or submucosal tissues of the extremities, larynx, face, abdomen, and genitals. Most cases of HAE are caused by mutations in the serpin family G member 1 gene (SERPING1), which encodes C1-esterase inhibitor (C1-INH) protein. Mutations in SERPING1 lead to deficient (type I HAE-C1-INH) or dysfunctional (type II HAE-C1-INH) C1-INH protein and subsequent dysregulation of the kallikrein–bradykinin cascade. However, some patients present with a third type of HAE (HAE-nI-C1-INH), which was first described in the year 2000 and is characterized by an absence of mutations in SERPING1. Although mutations in the coagulation factor XII, angiopoietin-1, plasminogen, kininogen-1, myoferlin, and heparan sulfate-glucosamine 3-O-sulfotransferase-6 genes have been identified in some patients with HAE-nI-C1-INH, genetic cause is still unknown in many cases, hindering full elucidation of the pathology of this HAE subtype. Diagnosis of HAE-nI-C1-INH is also further complicated by the fact that patients typically demonstrate normal plasma levels of C1-INH and complement component 4 protein and normal C1-INH functionality during laboratory analysis. Therefore, we review the challenges associated with diagnosing, treating, and living with HAE-nI-C1-INH. We conclude that raising awareness of the presenting features of HAE-nI-C1-INH within the clinical setting and among the general public is critical to aid earlier suspicion and diagnosis of the disease. Furthermore, adopting an individualized approach to HAE-nI-C1-INH treatment is essential to help address the current and significant unmet needs in this patient population.
Introduction
Hereditary angioedema (HAE) is a rare, chronic, and debilitating genetic disorder characterized by recurrent and unpredictable swelling episodes that primarily affect the subcutaneous and/or submucosal tissues of the extremities, larynx, face, abdomen, and genitals.Citation1–3 Most cases of HAE are caused by mutations in the serpin family G member 1 gene (SERPING1), leading to deficient (type I) or dysfunctional (type II) C1-esterase inhibitor (C1-INH) protein and subsequent dysregulation of the kallikrein–bradykinin cascade.Citation1–3 Dysregulation of the kallikrein–bradykinin cascade is associated with excessive production of bradykinin and consequent increases in vascular permeability resulting in episodes of edema.Citation1,Citation3,Citation4
Type I and type II HAE are collectively termed HAE-C1-INH, which is characterized by the detection of lower than normal plasma levels and/or functionality of the C1-INH protein and lower than normal plasma levels of complement component 4 (C4) in laboratory tests.Citation3,Citation4 However, some patients present with a third type of HAE, termed HAE-nI-C1-INH, which was first described in the year 2000 and is characterized by an absence of mutations in SERPING1.Citation5–7 Whereas it is documented in the literature that HAE-C1-INH has an estimated prevalence of 1 in 50,000 people,Citation1,Citation8 the prevalence, etiology, and pathophysiology of HAE-nI-C1-INH are less certain.Citation1,Citation9–11 Furthermore, the diagnosis of HAE-nI-C1-INH represents a significant challenge. This is in part because patients with HAE-nI-C1-INH typically demonstrate normal plasma levels of C1-INH and C4 proteins and normal C1-INH functionality during laboratory analysis.Citation3,Citation9
For the purpose of reliability, it is important to note that it is recommended that laboratory tests for C1-INH level and functionality are repeated one to three months later in patients with suspected HAE to confirm the results.Citation12 In addition, it has been reported in the literature that these laboratory tests can give rise to ambiguous results if patient blood samples are not handled carefully in such a way that decay of functional C1-INH is avoided prior to processing.Citation12–14 If such decay does occur prior to processing of a sample, this may consequently lead to a patient with HAE-nI-C1-INH being misdiagnosed with HAE-C1-INH (if sufficient additional tests are not performed), further complicating investigations into the true prevalence of HAE-nI-C1-INH.
Although mutations in the coagulation factor XII gene (FXII), angiopoietin-1 gene (ANGPT1), plasminogen gene (PLG), kininogen-1 gene (KNG1), myoferlin gene (MYOF), and heparan sulfate glucosamine 3-O-sulfotransferase-6 gene (HS3ST6) have been identified in some patients with HAE-nI-C1-INH, the genetic cause is still unknown in the majority of cases (see and for the proposed roles of these genes in the fibrinolytic system, the kallikrein/kinin system, and the development of angioedema).Citation6,Citation9,Citation13,Citation15–24 Additionally, mutations in FXII are estimated to account for only up to 25% of patients diagnosed with HAE-nI-C1-INH, and the prevalence of other known mutations in this patient population are less well characterized.Citation11,Citation22
Figure 1 Mutations in genes linked to some forms of HAE-nI-C1-INH and their roles within the fibrinolytic and kallikrein/kinin systems.Citation20,Citation21,Citation24,Citation25 Adapted from Bork K, Machnig T, Wulff K, Witzke G, Prusty S, Hardt J. Clinical features of genetically characterized types of hereditary angioedema with normal C1 inhibitor: a systematic review of qualitative evidence. Orphanet J Rare Dis. 2020;15(1):289. Creative Commons.Citation21
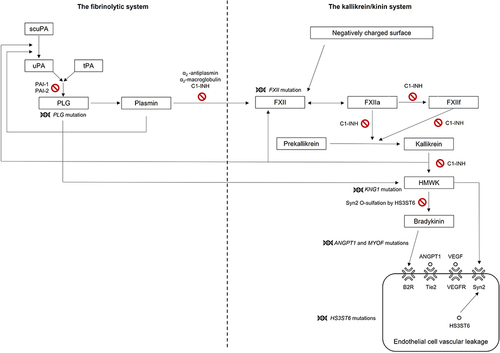
Table 1 Summary of Genes That Have Been Identified to Contain Mutations in Patients with HAE-nI-C1-INH and the Proposed Roles of Such Mutations in Angioedema
In a systematic review of the literature, patients with FXII HAE-nI-C1-INH were most frequently identified in Brazil, France, Spain, and Germany, with a smaller number of cases located in Turkey, Australia, Luxembourg, Morocco, and Italy.Citation21 Patients with PLG HAE-nI-C1-INH were more frequently identified in Germany, with a smaller number of cases located in Greece, Bulgaria, Spain, France, Italy, Japan, and the United States of America.Citation21 In the same systematic review, patients with ANGPT1 HAE-nI-C1-INH were identified in Italy, and patients with KNG1 HAE-nI-C1-INH were identified in Germany.Citation21 However, further investigation is required to obtain a clearer picture of the global distribution of clinical and genetic diagnoses of HAE-nI-C1-INH.
The purpose of this review is to raise further awareness of the clinical features of HAE-nI-C1-INH. The current unmet needs associated with diagnosis and treatment of HAE-nI-C1-INH will be highlighted, emphasizing the need for healthcare professionals to remain vigilant for potential cases of this form of HAE.
Diagnosis of HAE-nI-C1-INH
Clinical Presentation
HAE has a variable clinical course that is associated with multiple signs and symptoms of disease.Citation1 Hallmarks of both HAE-C1-INH and HAE-nI-C1-INH include recurrent angioedema that results in cutaneous and subcutaneous swelling, painful abdominal symptoms when the gastrointestinal tract is implicated, and respiratory symptoms when the airway is involved.Citation1
In contrast to the typical disease course of HAE-C1-INH, patients with HAE-nI-C1-INH usually experience their first symptoms of disease in late adolescence or early adulthood rather than during childhood.Citation1,Citation9,Citation27,Citation28 In a study of 138 patients with HAE-nI-C1-INH, the mean age of symptom onset was 26.8 years and only 8% of the patients experienced their first HAE attack before 10 years of age.Citation5,Citation29
Assessment of 295 patients with a confirmed diagnosis of HAE-nI-C1-INH indicated that the main known triggers for swelling attacks were hormones (68.3%), stress (59.6%), trauma (47.6%), and dental treatment (13.9%).Citation30 Patients with symptoms of HAE-nI-C1-INH are predominantly female, and exposure to estrogens (eg, during the use of oral contraceptives, hormone replacement therapy, and pregnancy) is often linked to exacerbation of the course of disease.Citation9,Citation21,Citation31–34 In a survey of 57 patients with FXII HAE-nI-C1-INH by the French National Center of Reference for Angioedema, exposure to estrogens was associated with HAE attacks in 95% (36/38) of symptomatic patients.Citation33,Citation35 In the same study, attacks were exacerbated during pregnancy and associated with the ingestion of estrogen-containing oral contraceptives in 67% (24/36) of symptomatic patients.Citation33,Citation35
Although the attacks experienced by patients with HAE-nI-C1-INH tend to look clinically similar to those that occur in patients with HAE-C1-INH, it is documented in the literature that swelling of the face, tongue, and oropharynx appear to occur more frequently in HAE-nI-C1-INH than in HAE-C1-INH.Citation1,Citation10,Citation11,Citation29,Citation36 A recent systematic literature review identified several clinical features of disease that appear to be more common in some forms of genetically identified HAE-nI-C1-INH versus other forms.Citation21 In this review, it was noted that estrogen use appears to have a more pronounced aggravating effect on HAE attacks in patients with HAE-nI-C1-INH with an FXII mutation versus those with HAE-nI-C1-INH due to other mutations.Citation21 In addition, the occurrence of hemorrhage at the swelling site appears to be more common in patients with HAE-nI-C1-INH with an FXII mutation versus those with HAE-nI-C1-INH due to other mutations.Citation21 Notably, a higher frequency of tongue swelling is reported in patients with HAE-nI-C1-INH with a PLG mutation versus those with HAE-nI-C1-INH due to other mutations and, in some instances, has been reported as the only clinical manifestation of the disease.Citation21 Erythema marginatum, which is common in HAE-C1-INH, was not observed in any of the HAE-nI-C1-INH cases captured in this systematic literature review.Citation21 Another study identified rare instances of joint pain that were independent of swelling episodes and unresponsive to immunosuppressant medications in patients with HAE-nI-C1-INH.Citation37
Prodromes are thought to occur in many patients with symptomatic HAE-C1-INH;Citation38,Citation39 however, further research is required to characterize them in patients with HAE-nI-C1-INH. Reports of prodromal fatigue and malaise have been documented in the literature for patients with HAE-nI-C1-INH as well as rare incidences of hemorrhages and/or bruising immediately before swelling becomes apparent.Citation40
Guidelines for Diagnosis
HAE can be differentiated from mast cell–mediated angioedema by several features. First, HAE is not associated with the formation of urticaria or pruritus.Citation1 Second, HAE swelling attacks tend to progress to maximum severity over the course of several hours and are generally longer lasting than the attacks associated with mast cell–mediated angioedema.Citation1 In support of this, reports in the literature indicate that symptoms of HAE can last for up to five days if left untreated.Citation3 Third, HAE attacks do not respond to common treatments that target mast cells, including antihistamines, corticosteroids, and epinephrines.Citation1
The United States Hereditary Angioedema Association Medical Advisory Board recommends that diagnosis of HAE-nI-C1-INH be based on the following clinical criteria:Citation1,Citation5,Citation41–43 the patient has a documented history of recurrent angioedema with no signs of concomitant urticaria and no use of medication that is known to cause angioedema, such as angiotensin-converting enzyme inhibitors;Citation44 laboratory testing indicates that the patient has normal, or close to normal, plasma levels of C4 and C1-INH antigens and normal C1-INH functionality; and the patient either has a genetic mutation in one of the genes known to be associated with HAE-nI-C1-INH or has a family history of recurrent angioedema episodes that were non-responsive to high-dose antihistamine therapy for at least one month or for a time interval that would be expected to be associated with three or more angioedema attacks (). Other supportive evidence for an HAE-nI-C1-INH diagnosis includes non-responsiveness to other mast cell–targeted therapies such as omalizumab, a history of rapid and durable responses to medications targeting the bradykinin pathway, and documented episodes of predominant and visible angioedema or, in patients with abdominal symptoms, documented evidence of edema in the bowel wall observed by computed tomography, ultrasound, or magnetic resonance imaging.Citation1,Citation5,Citation41–43 Similarly, the World Allergy Organization / European Academy of Allergy and Clinical Immunology guidelines for the management of HAE also recommend that a diagnosis of HAE-nI-C1-INH be confirmed through laboratory testing of C1 levels and function, genetic testing for known HAE-nI-C1-INH mutations, and family history.Citation13
Figure 2 Diagnostic algorithms for (a) HAE-C1-INH and (b) HAE-nI-C1-INH, based on the known clinical and genetic characteristics of the diseases. Presence of the supportive evidence shown in the boxes with blue outlines is not required for a confirmed diagnosis of HAE.Citation1,Citation45,Citation46
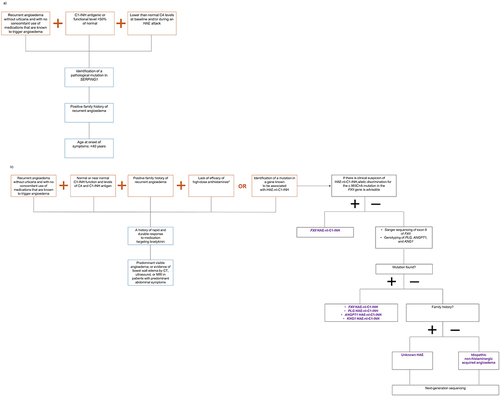
A recent algorithm has also been published in the literature to help clinicians further investigate patients with suspected HAE-nI-C1-INH.Citation45 According to this algorithm (), it would be beneficial for patients with suspected HAE-nI-C1-INH to undergo allelic screening for the c.983C>A mutation in FXII, regardless of whether they have a positive family history of the condition or not.Citation45 If this allelic screen is negative, then suggested further investigations include Sanger sequencing of exon 9 of FXII alongside genotyping of PLG, KNG1, and ANGPT1.Citation45 If mutations are not detected in these genes, the algorithm indicates that patients with a family history of the condition are more likely to have HAE-nI-C1-INH presenting as unknown HAE and patients without a family history are more likely to have idiopathic non-histaminergic acquired angioedema.Citation45 However, next-generation sequencing should be considered in both cases to improve diagnostic accuracy.Citation45
Challenges Associated with Obtaining a Confirmed Diagnosis
Findings from an International Patient Experience of HAE study indicate that 43% of 313 patients waited more than a year after their first swelling attack to seek medical help.Citation47,Citation48 In the same study, patients reported visiting an average of 4.4 physicians before receiving a confirmed diagnosis of HAE, with 65% receiving prior misdiagnoses.Citation47,Citation48
Providing a confirmed diagnosis of HAE-nI-C1-INH represents a substantial challenge for clinicians because there remains a significant unmet need for a validated biochemical test and/or biomarker that could specifically identify patients with this form of HAE.Citation1,Citation5 Furthermore, significant heterogeneity in clinical presentation and disease course among affected patients has been reported in the literature.Citation5
Although a positive family history of HAE can aid diagnosis, the rate of occurrence of de novo mutations in HAE-nI-C1-INH is currently unknown.Citation42 For comparative purposes, at least one-quarter of patients who receive a diagnosis of HAE-C1-INH are found to have de novo mutations and, therefore, no ancestral family history of the condition.Citation42,Citation49 Furthermore, the low penetrance of HAE-nI-C1-INH could result in a patient having affected family members who are asymptomatic and therefore undiagnosed.Citation5 In an Italian study that included 32 females with HAE-nI-C1-INH caused by mutations in FXII, 44.4% were asymptomatic.Citation34
Although genetic testing can help to provide a confirmed diagnosis, not all clinicians know about, or have access to, licensed laboratories that are able to use customized whole-exon sequencing processes to identify patients with mutations in genes that are confirmed to be associated with HAE-nI-C1-INH.Citation1,Citation42 In addition, some patients may present with clinical features and laboratory test results (eg, normal levels of C1-INH and C4) that point towards a diagnosis of HAE-nI-C1-INH in the absence of mutations in the genes that are currently known to be associated with the condition, thus hindering a conclusive diagnosis.Citation1 In these cases, a de novo mutation might be responsible for the patient’s symptoms; therefore, a diagnosis of HAE-nI-C1-INH cannot be completely ruled out.
Patients without a confirmed HAE diagnosis can experience significant difficulties obtaining insurance coverage for long-term treatment costs.Citation5,Citation50,Citation51 In many cases, treatment response to a trial course of therapy specific to HAE can form part of the diagnostic process for patients with HAE-nI-C1-INH and can help to justify the decision for consideration of long-term access to an on-demand medication and/or access to prophylactic treatment.Citation50,Citation52
Treatment of HAE-nI-C1-INH
Treatment Challenges
Failure to respond to treatment with corticosteroids and antihistamines frequently forms part of the diagnostic criteria for HAE-nI-C1-INH and is indicative of the absence of histamine degranulation and mast cell degranulation as pathological features of the disease.Citation1,Citation5 Early clinical evidence suggests that bradykinin might play an important role in many cases of HAE-nI-C1-INH, particularly in patients with HAE-nI-C1-INH due to mutations in FXII and PLG, but it is currently unclear whether it is implicated as the main mediator of edema in all forms of HAE-nI-C1-INH.Citation13,Citation24,Citation34,Citation53,Citation54 As a result, treatment of HAE-nI-C1-INH remains suboptimal, with further research required; however, it is important to emphasize that therapeutic options that have been approved for HAE-C1-INH should not be withheld from patients with HAE of unknown genetic cause in the meantime.Citation1
Often, discontinuation of exogenous estrogens is the first step in the treatment of women with suspected HAE-nI-C1-INH prior to initiation of any other therapies, including long-term prophylactics.Citation1,Citation36 Because of the absence of robust data from clinical trials and approved therapies specific to HAE-nI-C1-INH, treatment of patients is usually based on clinical experience with HAE-C1-INH and comprises on-demand (injectable ecallantide, injectable icatibant, and C1-INH concentrate infusion) and prophylactic (intravenous or subcutaneous C1-INH concentrate, injectable lanadelumab, and, more recently, oral berotralstat) agents that either directly or indirectly modulate the metabolism of bradykinin.Citation1,Citation5,Citation13,Citation34,Citation59–61 The proposed modes of action of these therapies in HAE-nI-C1-INH are shown in .
Table 2 Proposed Mode of Action of Treatments Offered to Patients with HAE-nI-C1-INH
In a recent systematic review, it was reported that on-demand and long-term prophylactic treatments of 43 patients with genetically confirmed HAE-nI-C1-INH commonly included the use of C1-INH concentrate, icatibant, progestins, and tranexamic acid.Citation21 Although examples of oral berotralstat use in patients with HAE-nI-C1-INH are currently limited in the literature, a case report of a patient with a severe needle phobia suggests that berotralstat can provide an effective option where the use of injectable lanadelumab and icatibant is less unfeasible.Citation50
In addition, findings from a recent retrospective case series of 23 patients indicate that the strategies used to treat patients with HAE-C1-INH, including prophylactic and on-demand therapies, may also be beneficial in patients with HAE-nI-C1-INH.Citation9 In this study, treatment regimens and outcomes were found to vary widely between patients, highlighting the need for personalized HAE-nI-C1-INH treatment plans.Citation9 Furthermore, in another study of six patients with confirmed HAE-nI-C1-INH, one patient responded well to tranexamic acid but not to C1-INH concentrate, four patients usually responded well to C1-INH concentrate, and one patient responded exclusively to icatibant during acute HAE attacks.Citation42 In addition, the importance of the availability of on-demand therapies for patients with HAE-nI-C1-INH was clearly demonstrated, even in individuals who were already receiving long-term prophylactic treatment.Citation9 It is crucial that both physicians and patients recognize the value of early treatment in preventing HAE attacks from progressing in severity.Citation1
It is important to consider that treatment response may also depend on the genetic subtype of HAE-nI-C1-INH that a patient presents with; therefore, a failed response to one therapeutic agent does not necessarily mean that HAE can be ruled out. In the literature, there are reports of attempts to customize treatment strategies in patients with genetically confirmed HAE-nI-C1-INH caused by mutations in either FXII or PLG.Citation21 Use of on-demand icatibant treatment by 13 patients with HAE-nI-C1-INH due to the c.988A>G mutation in PLG for a combined total of 201 acute episodes of facial swelling, abdominal swelling, and tongue swelling was shown to shorten the duration of attacks by 88%.Citation54 In the same study, use of plasma-derived C1-INH concentrate by 12 patients with HAE-nI-C1-INH due to the aforementioned mutation for a combined total of 74 acute episodes of facial, abdominal, and tongue swelling was shown to decrease the duration of attacks by 44%.Citation54 Furthermore, long-term prophylaxis resulted in a mean reduction in attack frequency of 93.9% with an antifibrinolytic (tranexamic acid; three patients), 83.3% with an attenuated androgen (danazol; three patients), and 46.3% with a progestin (desogestrel; six patients).Citation54 In contrast, corticosteroid and antihistamine use in these patients during acute attacks or for long-term prophylaxis resulted in a high number of non-responders.Citation54
In a separate study, 11 female patients with HAE-nI-C1-INH due to mutations in FXII who were treated with plasma-derived C1-INH concentrate for a total of 143 facial swelling episodes experienced reduced attack durations (mean ± standard deviation [SD] duration: 26.6 ± 10.1 hours) versus the durations of 88 prior untreated facial swelling episodes (mean ± SD duration: 64.1 ± 28.0 hours).Citation53 Long-term prophylaxis resulted in a mean reduction in attack frequency of 100% with danazol (three women), 99.8% with progestins after discontinuation of estrogen-containing oral contraceptives (16 women), and 93.8% with tranexamic acid (four women).Citation53
However, in a study of 21 patients with HAE-nI-C1-INH of unknown genetic cause who were taking tranexamic acid as a prophylactic treatment, only 11 experienced a consistent and persistent reduction in attack rate and 10 discontinued because of lack of efficacy, further highlighting the importance of a personalized approach to HAE therapy.Citation34 It is also important to note that without a biomarker or a biochemical test to help predict treatment response to medications that target the bradykinin pathway, this often means that both patients with confirmed HAE-nI-C1-INH and those with suspected HAE-nI-C1-INH can go through lengthy trials of various treatments before finding one that is right for them.Citation5,Citation9,Citation59
Pregnancy and HAE-nI-C1-INH Treatment
Therapeutic options for HAE are more limited during pregnancy and careful management of the patient is required.Citation7,Citation35 Experience of treating pregnant patients for HAE-nI-C1-INH attacks is limited to case reports and small case series in the literature.Citation35 Therefore, it is recommended that a management plan coordinated by a clinician who is specialized in HAE is put in place as early as possible after a patient becomes pregnant. Long-term prophylaxis of pregnant patients with HAE-nI-C1-INH has been described in the literature with no reported complications.Citation35,Citation62 Ideally, C1-INH concentrate should be made available at the patient’s maternity center and/or home so that treatment can be initiated as quickly as possible when an HAE attack begins.Citation35,Citation63
Comorbidities and HAE-nI-C1-INH Treatment
A recent population-based cohort study of 239 patients with HAE-C1-INH identified an increased risk of the following comorbidities versus a control cohort of 2383 individuals from the general population:Citation64 cardiovascular disease, including hypertension and arterial or venous thromboembolic disease; hyperlipidemia; and autoimmune diseases. Higher prescription rates for drugs targeting hypertension, hypothyroidism, and hyperlipidemia were also observed in patients with HAE versus the general population.Citation64
It is also documented that difficulties in obtaining a timely diagnosis of HAE, uncertainties around disease progression, and administration of treatment can result in anxiety and depression.Citation9,Citation65,Citation66 One survey study of 26 patients with HAE-C1-INH indicated that 39% and 15% of patients experienced depression and anxiety, respectively.Citation65
It is important to note that treatment choice can play a significant role in reducing the impact of anxiety and depression in patients with HAE. In a study of 37 patients with HAE, of whom four had been diagnosed with HAE-nI-C1-INH, it was demonstrated that patients who were receiving long-term prophylactic therapy had significantly lower scores for anxiety and depression versus those who were not.Citation66 Furthermore, in a recent case report, a patient with HAE-nI-C1-INH who declined a trial course of treatment with injectable lanadelumab and icatibant because of a severe needle phobia reported a significant improvement in anxiety levels when receiving prophylactic treatment with oral berotralstat.Citation50 This highlights the benefits of adopting a personalized approach to HAE management that takes into account a patient’s comorbidities and preferences.Citation50
Understanding comorbidities in patients with HAE is important because treatment for one condition may change the risk of another.Citation64 Use of attenuated androgens, such as danazol and oxandrolone, as prophylactic treatments can contribute to the comorbidities experienced by patients with HAE.Citation64 Long-term danazol use has been associated with atherogenic indices in the literature. Studies indicate that long-term androgen use may be associated with increased risks of hypertension, deep vein thrombosis, and liver cancer.Citation67–70
Although a full investigation of comorbidities in patients with HAE-nI-C1-INH has not yet been described in the literature, it is expected that these patients may experience a greater range of comorbidities because of older age at diagnosis and the psychological impact of managing a form of HAE that is less well studied. A recent study identified five cases of HAE-nI-C1-INH, three of which were complicated by epilepsy.Citation71 In one of these patients, administration of plasma-derived C1-INH concentrate during episodes of epileptic seizure and angioedema resulted in suppression of the seizures within 8 to 16 minutes, whereas medication specific to epilepsy had not been able to completely relieve the patient of their symptoms.Citation71 In another of the three patients, plasma-derived C1-INH concentrate and icatibant were effective at treating impaired consciousness during epileptic seizures.Citation71 Both cases underscore the impact that untreated HAE-nI-C1-INH can have on other comorbidities.Citation71 In contrast, no cases of epilepsy were identified in the 13 patients with HAE-C1-INH who were part of the same study, suggesting further investigation is required to determine if the prevalence of epilepsy is higher among patients with HAE-nI-C1-INH.Citation71
A case of pancreatitis has also been reported in the literature for a patient with HAE-nI-C1-INH caused by a mutation in FXII.Citation72 Although this patient did not receive any HAE-specific treatment to try to resolve the episode of pancreatitis, two other patients in the same study with HAE-C1-INH and pancreatitis responded well to treatment with icatibant, which is an antagonist of the bradykinin B2 receptor that has previously been shown to suppress induced pancreatitis.Citation72
Burden of Disease in Patients with HAE-nI-C1-INH
Because of the episodic nature of the disease, lack of awareness among healthcare professionals, lack of uniform diagnostic criteria, and a lack of approved therapeutics for HAE-nI-C1-INH, the burden of disease for patients remains significant.Citation5,Citation73
Burden of Disease Associated with Diagnostic Delays
It has been documented in the literature that all forms of HAE impose a significant burden on a patient’s quality of life, education, and work opportunities.Citation48 A combination of non-specific presenting symptoms and lack of a routinely available diagnostic test mean that many patients with HAE-nI-C1-INH face a series of prior misdiagnoses, which can cause significant disruption to daily life and lead to unnecessary surgical interventions.Citation5,Citation48,Citation74
During the time between a patient’s first HAE attack and receipt of a confirmed diagnosis of HAE, they may make multiple trips to physicians and emergency departments, which can result in loss of faith in a system that appears to be unable to diagnose or treat their condition.Citation48,Citation75 In a recent case series of 23 patients with HAE-nI-C1-INH, an average delay of greater than 10 years between the onset of HAE symptoms and receipt of a confirmed diagnosis was detected.Citation9 Lack of an accurate, early diagnosis can leave patients struggling with unexplained and unmanaged symptoms for prolonged periods of time, which can cause significant anxiety and depression.Citation9 These feelings of anxiety and depression can be worsened by the knowledge that upper airway swelling episodes can be potentially life-threatening if they are not treated appropriately and in a timely manner.Citation3,Citation5,Citation29
Abdominal pain is one of the most common symptoms of HAE, frequently leading to misdiagnosis of gastrointestinal disorders.Citation9,Citation72,Citation76 A retrospective study of 52 patients with normal C1-INH levels indicates that diagnoses of acute abdominal pain (n=16/52; 30.8%) and appendicitis (n=9/16; 56.2%) were frequently made. Furthermore, 81.2% of the patients diagnosed with acute abdominal pain underwent unnecessary invasive surgical procedures.Citation74 In support of this, a case series of 23 patients with HAE-nI-C1-INH indicates that a substantial percentage had a medical history of some form of abdominal surgery, including hysterectomy, oophorectomy, appendectomy, or cholecystectomy.Citation9
Burden of Disease Associated with Treatment
Adopting a personalized shared decision-making approach that takes patient preferences into account can be a valuable tool for managing the burden of treatment in patients with HAE. In a survey study of 149 patients with HAE-C1-INH, 76% reported having a personalized treatment plan for their condition versus only 50% of 37 patients with HAE-nI-C1-INH.Citation77 In addition, 88% of the 149 patients with HAE-C1-INH versus 76% of the 37 patients with HAE-nI-C1-INH reported having on-demand treatments available, and a smaller proportion of patients with HAE-nI-C1-INH (17%) reported receiving prophylactic therapy versus patients with HAE-C1-INH (66%). Notably, 85% of the patients with HAE-nI-C1-INH who participated in this study indicated that, in general, at least three-quarters of their attacks would be severe enough to adversely affect their quality of life if left untreated.Citation77
However, even with a treatment plan in place, the burden of treatment can be significant for patients with HAE.Citation78 Route of treatment administration can be an important consideration for many patients because it has a significant impact on both adherence to therapy and the perceived success of disease management.Citation52,Citation78 In a survey study of 75 patients with HAE-C1-INH, almost all users of prophylactic therapy indicated that, despite liking their current medication, they would prefer an oral option if available because they believed that this would be a better fit with their lifestyle versus an injectable treatment.Citation79 In this same study, 67% of users of prophylactic therapy (including both intravenous and subcutaneous therapies) agreed that avoiding needles was the primary reason they would try an oral prophylactic medicine.
There is also some early evidence in the literature that therapy may need to be trialed for longer in patients with HAE-nI-C1-INH versus in patients with HAE-C1-INH before treatment response is evaluated, which could exacerbate anxiety and frustration. For example, in a study of 22 patients with HAE-nI-C1-INH, the median time to resolution of swelling attacks following treatment with icatibant was found to be longer in patients with HAE-nI-C1-INH versus resolution in patients with type I HAE-C1-INH.Citation59 Therefore, more research and clinical trials are needed to determine optimum therapy regimens for patients with HAE-nI-C1-INH.
Although the development and approval of an array of therapeutics for HAE has helped to reduce the overall burden of disease; improve patient quality of life; and take some of the strain off urgent care, emergency departments, and hospitals, it is also important to note that the economic burden of acute and long-term therapies is high.Citation51 Specifically, concerns about the financial impact of these therapies on healthcare systems have resulted in the implementation of numerous barriers to and limitations on patient access to them.Citation51 Therefore, insurance coverage for treatment costs associated with the use of on-demand and prophylactic treatments for HAE is often dependent upon patients having a confirmed diagnosis.Citation50,Citation51
Burden of HAE-nI-C1-INH During Pregnancy
Evidence in the literature indicates that women diagnosed with HAE-C1-INH have a variable disease course during pregnancy, with hormonal changes not the only influential factor.Citation35,Citation80–82 A recent study of 45 pregnancies in 26 women diagnosed with HAE-nI-C1-INH indicates that the rate of spontaneous abortion (n=8/45; 17.8%) was comparable to that expected for women without HAE-nI-C1-INH.Citation35 However, HAE attacks occurred in 24 of 37 (64.7%) of the pregnancies and were more frequent in the first trimester (41.7%) versus the second (12.5%) and third (20.8%) trimesters.Citation35 Furthermore, of the 15 patients who experienced HAE attacks both before and during their first pregnancy, 9 (60%) reported a worsening in attack frequency. Therefore, pregnancy may worsen the course of HAE-nI-C1-INH disease for some patients, causing significant levels of anxiety. This anxiety may be further exacerbated by the knowledge that obstetrical complications need to be ruled out at the time of attackCitation35,Citation63 and, as discussed above, therapeutic options for HAE are limited during pregnancy.Citation35,Citation63 In addition, patients may encounter challenging disparities in healthcare resources, including restrictions on the availability of plasma-derived C1-INH concentrate (which has some dependence on geographical location) and barriers to obtaining insurance coverage for the cost of treatment, meaning treatment access could be dependent upon the patient’s financial situation in some cases.Citation35,Citation63,Citation83
Recommendations for Diagnosis and Treatment of HAE-nI-C1-INH
Based on the literature reviewed in this article, we recommend following the diagnostic algorithm proposed by the US Hereditary Angioedema Association.Citation1 Where able, we also recommend pursuing genetic testing as described in . The importance of encouraging physicians to undertake and their patients to undergo genetic testing when HAE-nI-C1-INH is suspected should not be underestimated, since this will aid the reliability of diagnosis and contribute to further understanding of the etiology of the disease. However, we recognize the need for a clinical diagnosis should genetic testing be unavailable or inconclusive.
Effective treatment of HAE-nI-C1-INH requires trial and error of recommended medications, including those approved by the US Food and Drug Administration for HAE-C1-INH. Importantly, the use of some medications to treat HAE-nI-C1-INH may be off-label. Particular attention should be given to the duration of the treatment trial with high-dose antihistamines. A lack of efficacy of high-dose antihistamine therapy should be documented for at least one month or for an interval that is expected to be associated with three or more angioedema attacks, which might exceed one month for some patients. We recommend trying more than one of the treatments that are used for mast cell–mediated angioedema, including omalizumab. In patients with a confirmed diagnosis of factor XII mutation, tranexamic acid could be trialed. Comorbidities associated with the treatment of HAE-nI-C1-INH should be appropriately monitored and managed using a shared decision-making approach between the physician and the patient wherever possible. Comorbidities that are specifically associated with the disease course of HAE-nI-C1-INH should also be regularly reviewed and discussed with patients to enable implementation of an individualized treatment plan that can successfully aid optimization of quality of life.
Conclusions
The prevalence of HAE-nI-C1-INH remains unclear, with difficulties in diagnosis and the lack of a confirmed and specific biochemical test and/or biomarker highlighting the need for healthcare professionals to remain vigilant for potential cases of this form of HAE. Although current research suggests that most of the forms of HAE-nI-C1-INH that have been genetically identified to date may be due to different biochemical disturbances in the kallikrein/kinin and fibrinolytic systems, further research is required to fully elucidate the pathological mechanisms of the disease. Therefore, raising awareness of the presenting features of HAE-nI-C1-INH within both the clinical setting and the general public is critical to aid earlier suspicion and diagnosis of the disease. Furthermore, adopting a personalized approach to HAE-nI-C1-INH treatment is essential to help address the current and significant unmet needs in this patient population.
Abbreviations
ANGPT1, angiopoietin-1; ANGPT1, angiopoietin-1 gene; B2R, bradykinin-2 receptor; C1-INH, C1-esterase inhibitor; C4, complement component 4; CT, computed tomography; FDA, Food and Drug Administration; FXII, coagulation factor XII; FXII, coagulation factor XII gene; FXIIa, activated coagulation factor XII; FXIIf, coagulation factor XII fragment; HAE, hereditary angioedema; HAE-C1-INH, hereditary angioedema due to C1-esterase inhibitor deficiency or dysfunction; HAE-nI-C1-INH, hereditary angioedema with normal C1-esterase inhibitor levels; HMWK, high molecular weight kininogen; HS3ST6, heparan sulfate glucosamine 3-O-sulfotransferase-6; HS3ST6, heparan sulfate glucosamine 3-O-sulfotransferase-6 gene; KNG1, kininogen-1; KNG1, kininogen-1 gene; MRI, magnetic resonance imaging; MYOF, myoferlin; MYOF, myoferlin gene; PAI, plasminogen activator inhibitor; PLG, plasminogen; PLG, plasminogen gene; ScuPA, single-chain urokinase-type plasminogen activator; SD, standard deviation; SERPING1, serpin family G member 1 gene; Syn2, syndecan-2; Tie2, tyrosine-protein kinase; tPA, tissue plasminogen activator; uPA, urokinase-type plasminogen activator; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor.
Consent for Publication
All authors agreed to publication of this work.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
Douglas Jones reports being a consultant for and receiving consulting fees from AstraZeneca, BioCryst, Pharming Technologies BV, Pharvaris, Sanofi-Regeneron, and Takeda Pharmaceutical Company Ltd. He also participates in the medical advisory board for KalVista. Heidi Zafra is a member of the BioCryst Advisory Board, and the Takeda Advisory Board. John Anderson affiliates with AllerVie Health and is a speaker bureau member for CSL Behring, Pharming, BioCryst, and Takeda; and has received consulting fees from and is a clinical trial investigator for BioCryst, CSL Behring, Pharming, Takeda, KalVista, Pharvaris, and BioMarin, and consultation fees from Cycle Pharmaceuticals. The authors report no other conflicts of interest in this work.
Acknowledgments
Medical writing support was provided by Jennifer Shepherd, PhD, of Porterhouse Medical Group under the direction of the authors and in line with GPP3 guidelines.
Additional information
Funding
References
- Busse PJ, Christiansen SC, Riedl MA, et al. US HAEA medical advisory board 2020 guidelines for the management of hereditary angioedema. J Allergy Clin Immunol Pract. 2021;9(1):132–150.e3. doi:10.1016/j.jaip.2020.08.046
- Caballero T. Treatment of hereditary angioedema. J Investig Allergol Clin Immunol. 2021;31(1):1–16. doi:10.18176/jiaci.0653
- Longhurst HJ, Bork K. Hereditary angioedema: an update on causes, manifestations and treatment. Br J Hosp Med. 2019;80(7):391–398. doi:10.12968/hmed.2019.80.7.391
- Banday AZ, Kaur A, Jindal AK, Rawat A, Singh S. An update on the genetics and pathogenesis of hereditary angioedema. Genes Dis. 2019;7(1):75–83. doi:10.1016/j.gendis.2019.07.002
- Zuraw BL, Bork K, Binkley KE, et al. Hereditary angioedema with normal C1 inhibitor function: consensus of an international expert panel. Allergy Asthma Proc. 2012;33(Suppl 1):S145–S156. doi:10.2500/aap.2012.33.3627
- Bork K, Barnstedt SE, Koch P, Traupe H. Hereditary angioedema with normal C1-inhibitor activity in women. Lancet. 2000;356(9225):213–217. doi:10.1016/S0140-6736(00)02483-1
- Binkley KE, Davis A. Clinical, biochemical, and genetic characterization of a novel estrogen-dependent inherited form of angioedema. J Allergy Clin Immunol. 2000;106(3):546–550. doi:10.1067/mai.2000.108106
- Busse PJ, Christiansen SC. Hereditary angioedema. N Engl J Med. 2020;382(12):1136–1148. doi:10.1056/NEJMra1808012
- Jones DH, Bansal P, Bernstein JA, et al. Clinical profile and treatment outcomes in patients with hereditary angioedema with normal C1 esterase inhibitor. World Allergy Organ J. 2022;15(1):100621. doi:10.1016/j.waojou.2021.100621
- Riedl MA. Hereditary angioedema with normal C1-INH (HAE type III). J Allergy Clin Immunol Pract. 2013;1(5):427–432. doi:10.1016/j.jaip.2013.06.004
- Magerl M, Germenis AE, Maas C, Maurer M. Hereditary angioedema with normal C1 inhibitor: update on evaluation and treatment. Immunol Allergy Clin North Am. 2017;37(3):571–584. doi:10.1016/j.iac.2017.04.004
- Henao MP, Kraschnewski JL, Kelbel T, Craig TJ. Diagnosis and screening of patients with hereditary angioedema in primary care. Ther Clin Risk Manag. 2016;12:701–711. doi:10.2147/TCRM.S86293
- Maurer M, Magerl M, Betschel S, et al. The international WAO/EAACI guideline for the management of hereditary angioedema - the 2021 revision and update. World Allergy Organ J. 2022;15(3):100627. doi:10.1016/j.waojou.2022.100627
- Gompels MM, Lock RJ, Unsworth DJ, Johnston SL, Archer CB, Davies SV. Misdiagnosis of hereditary angio-oedema type 1 and type 2. Br J Dermatol. 2003;148(4):719–723. doi:10.1046/j.1365-2133.2003.05231.x
- Dewald G, Bork K. Missense mutations in the coagulation factor XII (Hageman factor) gene in hereditary angioedema with normal C1 inhibitor. Biochem Biophys Res Commun. 2006;343(4):1286–1289. doi:10.1016/j.bbrc.2006.03.092
- Bafunno V, Firinu D, D’Apolito M, et al. Mutation of the angiopoietin-1 gene (ANGPT1) associates with a new type of hereditary angioedema. J Allergy Clin Immunol. 2018;141(3):1009–1017. doi:10.1016/j.jaci.2017.05.020
- Bork K, Wulff K, Steinmüller‐Magin L, et al. Hereditary angioedema with a mutation in the plasminogen gene. Allergy. 2018;73(2):442–450. doi:10.1111/all.13270
- Bork K, Wulff K, Rossmann H, et al. Hereditary angioedema cosegregating with a novel kininogen 1 gene mutation changing the N‐terminal cleavage site of bradykinin. Allergy. 2019;74(12):2479–2481. doi:10.1111/all.13869
- Ariano A, D’Apolito M, Bova M, et al. A myoferlin gain-of-function variant associates with a new type of hereditary angioedema. Allergy. 2020;75(11):2989–2992. doi:10.1111/all.14454
- Bork K, Wulff K, Möhl BS, et al. Novel hereditary angioedema linked with a heparan sulfate 3-O-sulfotransferase 6 gene mutation. J Allergy Clin Immunol. 2021;148(4):1041–1048. doi:10.1016/j.jaci.2021.01.011
- Bork K, Machnig T, Wulff K, Witzke G, Prusty S, Hardt J. Clinical features of genetically characterized types of hereditary angioedema with normal C1 inhibitor: a systematic review of qualitative evidence. Orphanet J Rare Dis. 2020;15(1):289. doi:10.1186/s13023-020-01570-x
- Loules G, Parsopoulou F, Zamanakou M, et al. Deciphering the genetics of primary angioedema with normal levels of C1 inhibitor. J Clin Med. 2020;9(11):3402. doi:10.3390/jcm9113402
- Sharma J, Jindal AK, Banday AZ, et al. Pathophysiology of hereditary angioedema (HAE) beyond the SERPING1 gene. Clin Rev Allergy Immunol. 2021;60(3):305–315. doi:10.1007/s12016-021-08835-8
- Dickeson SK, Kumar S, Sun MF, et al. A mechanism for hereditary angioedema caused by a lysine 311-to-glutamic acid substitution in plasminogen. Blood. 2022;139(18):2816–2829. doi:10.1182/blood.2021012945
- Napolitano F, Montuori N. The role of the plasminogen activation system in angioedema: novel insights on the pathogenesis. J Clin Med. 2021;10(3):518. doi:10.3390/jcm10030518
- d’Apolito M, Santacroce R, Colia AL, Cordisco G, Maffione AB, Margaglione M. Angiopoietin‐1 haploinsufficiency affects the endothelial barrier and causes hereditary angioedema. Clin Exp Allergy. 2019;49(5):626–635. doi:10.1111/cea.13349
- Farkas H, Martinez‐Saguer I, Bork K, et al. International consensus on the diagnosis and management of pediatric patients with hereditary angioedema with C1 inhibitor deficiency. Allergy. 2017;72(2):300–313. doi:10.1111/all.13001
- Salguero CAS, Chacon AIS. HAE in children- what is the best treatment strategy? Int J Aller Medications. 2016;2(1):16. doi:10.23937/2572-3308.1510016
- Bork K, Gül D, Hardt J, Dewald G. Hereditary angioedema with normal C1 inhibitor: clinical symptoms and course. Am J Med. 2007;120(11):987–992. doi:10.1016/j.amjmed.2007.08.021
- Fragnan NTML, Veronez C, Moreno A, et al. Treatment of patients with hereditary angioedema with normal C1 inhibitor: evaluation of 295 patients. J Allergy Clin Immunol. 2019;143(2):AB40. doi:10.1016/j.jaci.2018.12.120
- Marcos C, López Lera A, Varela S, Liñares T, Alvarez-Eire MG, López-Trascasa M. Clinical, biochemical, and genetic characterization of type III hereditary angioedema in 13 Northwest Spanish families. Ann Allergy Asthma Immunol. 2012;109(3):195–200.e2. doi:10.1016/j.anai.2012.05.022
- Bork K, Wulff K, Hardt J, Witzke G, Staubach P. Hereditary angioedema caused by missense mutations in the factor XII gene: clinical features, trigger factors, and therapy. J Allergy Clin Immunol. 2009;124(1):129–134. doi:10.1016/j.jaci.2009.03.038
- Deroux A, Boccon-Gibod I, Fain O, et al. Hereditary angioedema with normal C1 inhibitor and factor XII mutation: a series of 57 patients from the French National Center of Reference for Angioedema. Clin Exp Immunol. 2016;185(3):332–337. doi:10.1111/cei.12820
- Bova M, Suffritti C, Bafunno V, et al. Impaired control of the contact system in hereditary angioedema with normal C1‐inhibitor. Allergy. 2020;75(6):1394–1403. doi:10.1111/all.14160
- Gabriel N, Marcelino F, Ferriani MPL, et al. Pregnancy in patients with hereditary angioedema and normal C1 inhibitor. Front Allergy. 2022;3:846968. doi:10.3389/falgy.2022.846968
- Bork K. Diagnosis and treatment of hereditary angioedema with normal C1 inhibitor. Allergy Asthma Clin Immunol. 2010;6(1):15. doi:10.1186/1710-1492-6-15
- Jones D, Park N, Thompson A. M101 joint pain: a rare symptom in patients with hereditary angioedema with normal C1-INH. Ann Allergy Asthma Immunol. 2021;127(5 Suppl):S82. doi:10.1016/j.anai.2021.08.255
- Caballero T, Maurer M, Longhurst HJ, et al. Triggers and prodromal symptoms of angioedema attacks in patients with hereditary angioedema. J Investig Allergol Clin Immunol. 2016;26(6):383–386. doi:10.18176/jiaci.0102
- Magerl M, Doumoulakis G, Kalkounou I, et al. Characterization of prodromal symptoms in a large population of patients with hereditary angio‐oedema. Clin Exp Dermatol. 2014;39(3):298–303. doi:10.1111/ced.12285
- Taya J, Veronez CL, Pesquero JB, Bork K, Grumach AS. Uncommon signs associated with hereditary angioedema with normal C1 inhibitor. J Investig Allergol Clin Immunol. 2021;31(3):257–258. doi:10.18176/jiaci.0509
- Lara-Marquez ML, Christiansen SC, Riedl MA, Herschbach J, Zuraw BL. Threshold-stimulated kallikrein activity distinguishes bradykinin- from histamine-mediated angioedema. Clin Exp Allergy. 2018;48(11):1429–1438. doi:10.1111/cea.13219
- McKibbin L, Barber C, Kalicinsky C, Warrington R. Review of the Manitoba cohort of patients with hereditary angioedema with normal C1 inhibitor. Allergy Asthma Clin Immunol. 2019;15:66. doi:10.1186/s13223-019-0381-y
- Bernstein JA. Severity of hereditary angioedema, prevalence, and diagnostic considerations. Am J Manag Care. 2018;24(14 Suppl):S292–S298.
- Zafra H. Hereditary angioedema: a review. WMJ. 2022;121(1):48–53.
- Dias MM, Moreno AS, Maia LSM, et al. A cost-effective algorithm for diagnosis of hereditary angioedema with normal C1 inhibitor: applying molecular approach to clinical practice. J Allergy Clin Immunol Pract. 2020;8(1):419–421.e4. doi:10.1016/j.jaip.2019.06.041
- Cicardi M, Zuraw BL. Angioedema due to bradykinin dysregulation. J Allergy Clin Immunol Pract. 2018;6(4):1132–1141. doi:10.1016/j.jaip.2018.04.022
- Lunn ML, Santos CB, Craig TJ. Is there a need for clinical guidelines in the United States for the diagnosis of hereditary angioedema and the screening of family members of affected patients? Ann Allergy Asthma Immunol. 2010;104(3):211–214. doi:10.1016/j.anai.2009.12.004
- Lumry WR, Settipane RA. Hereditary angioedema: epidemiology and burden of disease. Allergy Asthma Proc. 2020;41(Suppl 1):S08–S13. doi:10.2500/aap.2020.41.200050
- Pappalardo E, Cicardi M, Duponchel C, et al. Frequent de novo mutations and exon deletions in the C1inhibitor gene of patients with angioedema. J Allergy Clin Immunol. 2000;106(6):1147–1154. doi:10.1067/mai.2000.110471
- Kelbel T. A case of normal C1 esterase inhibitor hereditary angioedema successfully treated with berotralstat. Ann Allergy Asthma Immunol. 2022;128(4):462–463. doi:10.1016/j.anai.2022.01.014
- Lumry WR. Hereditary angioedema: the economics of treatment of an orphan disease. Front Med. 2018;5:22. doi:10.3389/fmed.2018.00022
- Riedl MA, Banerji A, Gower R. Current medical management of hereditary angioedema: follow-up survey of US physicians. Ann Allergy Asthma Immunol. 2021;126(3):264–272. doi:10.1016/j.anai.2020.10.009
- Bork K, Wulff K, Witzke G, Hardt J. Treatment for hereditary angioedema with normal C1‐INH and specific mutations in the F12 gene (HAE‐FXII). Allergy. 2017;72(2):320–324. doi:10.1111/all.13076
- Bork K, Wulff K, Witzke G, Machnig T, Hardt J. Treatment of patients with hereditary angioedema with the c.988A>G (p.Lys330Glu) variant in the plasminogen gene. Orphanet J Rare Dis. 2020;15(1):52. doi:10.1186/s13023-020-1334-8
- Perego F, Wu MA, Valerieva A, et al. Current and emerging biologics for the treatment of hereditary angioedema. Expert Opin Biol Ther. 2019;19(6):517–526. doi:10.1080/14712598.2019.1595581
- Saule C, Boccon‐Gibod I, Fain O, et al. Benefits of progestin contraception in non‐allergic angioedema. Clin Exp Allergy. 2013;43(4):475–482. doi:10.1111/cea.12055
- Crosignani P, Olive D, Bergqvist A, Luciano A. Advances in the management of endometriosis: an update for clinicians. Hum Reprod Update. 2006;12(2):179–189. doi:10.1093/humupd/dmi049
- Luciano AA, Turksoy RN, Carleo J. Evaluation of oral medroxyprogesterone acetate in the treatment of endometriosis. Obstet Gynecol. 1988;72(3 Pt 1):323–327.
- Bouillet L, Boccon‐Gibod I, Launay D, et al. Hereditary angioedema with normal C1 inhibitor in a French cohort: clinical characteristics and response to treatment with icatibant. Immun Inflamm Dis. 2017;5(1):29–36. doi:10.1002/iid3.137
- ORLADEYO™ (berotralstat) capsules, for oral use [prescribing information]. Durham, NC, USA: BioCryst Pharmaceuticals, Inc; 2020.
- European Medicines Agency. Summary of opinion (initial authorisation): Orladeyo: berotralstat; 2021. Available from: https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-orladeyo_en.pdf. Accessed October 7, 2022.
- Garcia JFB, Takejima P, Veronez CL, et al. Use of pdC1-INH concentrate for long-term prophylaxis during pregnancy in hereditary angioedema with normal C1-INH. J Allergy Clin Immunol Pract. 2018;6(4):1406–1408. doi:10.1016/j.jaip.2017.12.022
- Caballero T, Canabal J, Rivero-Paparoni D, Cabañas R. Management of hereditary angioedema in pregnant women: a review. Int J Womens Health. 2014;6:839–848. doi:10.2147/IJWH.S46460
- Sundler Björkman L, Persson B, Aronsson D, Skattum L, Nordenfelt P, Egesten A. Comorbidities in hereditary angioedema-A population‐based cohort study. Clin Transl Allergy. 2022;12(3):e12135. doi:10.1002/clt2.12135
- Fouche AS, Saunders EFH, Craig T. Depression and anxiety in patients with Hereditary angioedema. Ann Allergy Asthma Immunol. 2014;112(4):371–375. doi:10.1016/j.anai.2013.05.028
- Zarnowski J, Rabe M, Kage P, Simon JC, Treudler R. Prophylactic treatment in hereditary angioedema is associated with reduced anxiety in patients in Leipzig, Germany. Int Arch Allergy Immunol. 2021;182(9):819–826. doi:10.1159/000514973
- Bork K, Bygum A, Hardt J. Benefits and risks of danazol in hereditary angioedema: a long-term survey of 118 patients. Ann Allergy Asthma Immunol. 2008;100(2):153–161. doi:10.1016/S1081-1206(10)60424-3
- Birjmohun RS, Kees Hovingh G, Stroes ESG, et al. Effects of short-term and long-term danazol treatment on lipoproteins, coagulation, and progression of atherosclerosis: two clinical trials in healthy volunteers and patients with Hereditary angioedema. Clin Ther. 2008;30(12):2314–2323. doi:10.1016/j.clinthera.2008.12.021
- Füst G, Farkas H, Csuka D, Varga L, Bork K. Long‐term efficacy of danazol treatment in hereditary angioedema. Eur J Clin Invest. 2011;41(3):256–262. doi:10.1111/j.1365-2362.2010.02402.x
- Kalaria S, Craig T. Assessment of hereditary angioedema treatment risks. Allergy Asthma Proc. 2013;34(6):519–522. doi:10.2500/aap.2013.34.3702
- Kuwahara S, Fukunaga A, Ohata M, et al. High prevalence of epilepsy in HAE with normal C1-INH. Allergol Int. 2020;69(4):630–632. doi:10.1016/j.alit.2020.04.008
- Veronez CL, Campos RA, Constantino-Silva RN, Nicolicht P, Pesquero JB, Grumach AS. Hereditary angioedema-associated acute pancreatitis in C1-inhibitor deficient and normal C1-inhibitor patients: case reports and literature review. Front Med. 2019;6:80. doi:10.3389/fmed.2019.00080
- Bygum A. Hereditary angio-oedema for dermatologists. Dermatology. 2019;235(4):263–275. doi:10.1159/000500196
- Gutierrez M, Veronez CL, Rodrigues Valle SO, et al. Unnecessary abdominal surgeries in attacks of hereditary angioedema with normal C1 inhibitor. Clin Rev Allergy Immunol. 2021;61(1):60–65. doi:10.1007/s12016-021-08852-7
- Malesker M. Addressing the individualized needs in hereditary angioedema: managed care strategies to optimize access to care. Am J Manag Care. 2022;28(1 Suppl):S3–S9. doi:10.37765/ajmc.2022.88822
- Veronez CL, Moreno AS, Constantino-Silva RN, et al. Hereditary angioedema with normal C1 inhibitor and F12 mutations in 42 Brazilian families. J Allergy Clin Immunol Pract. 2018;6(4):1209–1216.e8. doi:10.1016/j.jaip.2017.09.025
- Banerji A, Busse P, Christiansen SC, et al. Current state of hereditary angioedema management: a patient survey. Allergy Asthma Proc. 2015;36(3):213–217. doi:10.2500/aap.2015.36.3824
- Maurer M, Aygören-Pürsün E, Banerji A, et al. Consensus on treatment goals in hereditary angioedema: a global Delphi initiative. J Allergy Clin Immunol. 2021;148(6):1526–1532. doi:10.1016/j.jaci.2021.05.016
- Geba D, Mohd Sani J, Gascon M, Hahn R, Aggarwal K, Rosselli J. Hereditary angioedema patients would prefer newer-generation oral prophylaxis. J Drug Assess. 2021;10(1):51–56. doi:10.1080/21556660.2020.1863699
- Martinez-Saguer I, Rusicke E, Aygören-Pürsün E, Heller C, Klingebiel T, Kreuz W. Characterization of acute hereditary angioedema attacks during pregnancy and breast-feeding and their treatment with C1 inhibitor concentrate. Am J Obstet Gynecol. 2010;203(2):131.e1–131.e7. doi:10.1016/j.ajog.2010.03.003
- Czaller I, Visy B, Csuka D, Füst G, Tóth F, Farkas H. The natural history of hereditary angioedema and the impact of treatment with human C1-inhibitor concentrate during pregnancy: a long-term survey. Eur J Obstet Gynecol Reprod Biol. 2010;152(1):44–49. doi:10.1016/j.ejogrb.2010.05.008
- Machado AM, Pires RM, Martins RO, Grumach AS. Pregnancy and postpartum in hereditary angioedema with C1 inhibitor deficit in women who have no access to therapy. J Investig Allergol Clin Immunol. 2017;27(5):322–323. doi:10.18176/jiaci.0175
- Giavina-Bianchi P, Arruda LK, Aun MV, et al. Brazilian guidelines for hereditary angioedema management - 2017 update part 1: definition, classification and diagnosis. Clinics. 2018;73:e310. doi:10.6061/clinics/2018/e310