
Abstract
Multicentric Castleman’s disease (MCD), a distinct subtype of Castleman’s disease, is a rare, nonneoplastic, lymphoproliferative disorder. Patients with MCD present with systemic symptoms and multiple lymphadenopathy. Lymph node biopsy is necessary for the diagnosis of various histological MCD patterns including hyaline vascular, plasma cell, and mixed types. Human herpesvirus 8 (HHV8) infection was identified as an important etiology of MCD among immunocompromised patients such as those positive for human immunodeficiency virus. Although HHV8-negative MCD was reported in immunocompetent patients, the underlying etiology remains unknown. Several experts speculate that MCD in immunocompetent patients might be due to proinflammatory hypercytokinemia because of infection by a virus other than HHV8, inflammation, or neoplastic disease. In 2010, a distinct variant of HHV8-negative MCD reported in Japan was characterized by thrombocytopenia, anasarca, myelofibrosis, renal dysfunction, and organomegaly (TAFRO). Recent case reports and a systematic review suggest that TAFRO syndrome might have a unique pathogenesis among HHV8-negative MCD variants. This review introduces TAFRO syndrome as a subtype of HHV8-negative MCD and offers an overview of the current perspectives on this syndrome.
Introduction
Castleman’s disease (CD), first reported in 1954, is a rare, nonneoplastic, lymphoproliferative disorder.Citation1 Characteristic histopathological findings of CD are angiofollicular lymph node hyperplasia in a localized lymph node region.Citation2 In 1974, Gaba et al described cases exhibiting this distinct histopathology in multiple lymph nodes and clinically classified CD into unicentric CD (UCD) and multicentric CD (MCD).Citation3 Patients with UCD, who are usually younger than those with MCD, are often asymptomatic. Physical findings of UCD consist of enlargement of one lymph node with no radiologic evidence of lymphadenopathy in other areas. Conversely, patients with MCD have histologic findings of CD within at least one regional group of lymph nodes as well as clinical or radiologic evidence of additional lymphadenopathy. Patients with MCD show systemic symptoms stemming from reactive proliferation of benign lymphocytes. UCD is treated by surgical lymph node excision, which is curative for most patients. In contrast, systemic treatment is needed to control MCD.Citation4,Citation5 MCD is further classified according to the patient’s human herpesvirus 8 (HHV8) infection status. The histopathology of CD is classified into four variants: hyaline vascular (HV), plasma cell (PC), mixed, and plasmablastic.Citation5,Citation6 The plasmablastic variant is found only in patients with HHV8-associated MCD. Histological features of the HV variant include widened mantle zones composed of concentric rings of small lymphocytes in an onion-skin pattern around small atrophic germinal centers with penetrating hyalinized vessels and dysplastic follicular dendritic cells (FDCs).Citation7 In 2010, Takai et al proposed a new variant of HHV8-negative MCD characterized by thrombocytopenia, anasarca, myelofibrosis, renal dysfunction, and organomegaly and termed these presentations as thrombocytopenia, anasarca, myelofibrosis, renal dysfunction, and organomegaly (TAFRO) syndrome.Citation8 We herein review TAFRO syndrome as a novel clinical entity in HHV8-negative MCD patients, which represents a group of systemic inflammatory disorders derived from an autoimmune pathophysiology.
Clinical and histopathological features of MCD
MCD is a distinct subtype of CD presenting with multiple lesions and systemic symptoms. Clinical features of MCD are systemic inflammation, reactive proliferation of benign lymphocytes, multicentric lymphadenopathy, polyclonal hypergammaglobulinemia, microcytic anemia, hypoalbuminemia, and elevated serum inflammatory proteins such as C-reactive protein (CRP).Citation7,Citation9 MCD diagnosis is histologically confirmed by lymph node biopsy. These clinical and histopathological abnormalities might reflect hypercytokinemia based on unknown etiologies.
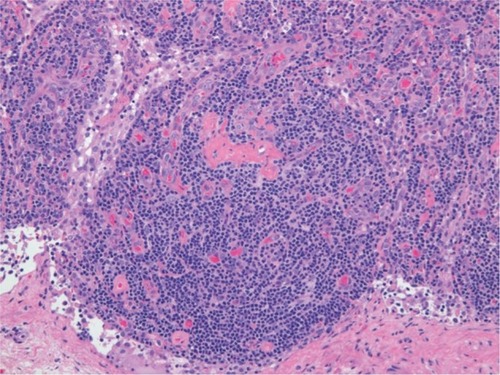
Lymph node enlargement with distinctive histological features characterizes MCD. As with CD, the histological patterns of MCD include HV, PC, and mixed types. The HV type is the more common MCD variant, whereas the PC type mostly involves mediastinum.Citation6 However, all lymph node-bearing regions and even tissues other than lymph nodes can be involved. The HV type is characterized by the proliferation of small, morphologically distinctive follicles that obscure the underlying nodal architecture (). The follicles are surrounded by roughly concentric circular layers resembling onion skin which are composed of small lymphocytes in the mantle zone, with circularly arranged capillaries penetrating the germinal centers;Citation10 these structures contain hyaline deposits and display a characteristic whorled appearance. In contrast, the interfollicular stromal tissue demonstrates prominent proliferation of capillaries and contains many small lymphocytes and PCs. Germinal centers are more typical of reactive follicular hyperplasia.
Figure 1 Histopathological features of hyaline-vascular type in a patient with multicentric Castleman’s disease. Left inguinal node biopsy shows atrophic germinal centers and expansion of the interfollicular zone with vascular proliferation (hematoxylin and eosin stain).
HHV8 status and current classification of MCD
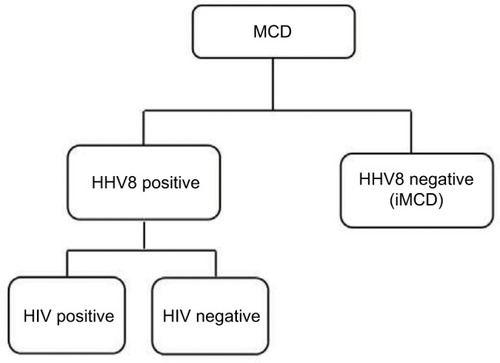
Human immunodeficiency virus (HIV) infection has been recognized as an important risk factor for MCD, and all patients with HIV-associated MCD are coinfected with HHV8. Approximately 50% of HIV-negative MCD cases are infected with HHV8, which varies with the HHV8 prevalence in the population.Citation11 Among patients with HHV8-associated MCD, HIV infection or another cause of immune compromise enables HHV8 to replicate in lymph nodes and to release human interleukin (IL)-6 and several other proinflammatory factors derived from HHV8.Citation12,Citation13 These proinflammatory factors induce proliferation of B lymphocytes and PCs, secretion of vascular endothelial growth factor (VEGF), which promotes angiogenesis, and an acute phase reaction.Citation14 Plasma titers of HHV8 correlate with clinical symptoms and predict relapse rates in HIV-associated MCD.Citation15 HHV8 infection is responsible for MCD etiology in both HIV-positive and HIV-negative patients. Therefore, traditional HIV-based classification is not appropriate for identifying factors driving MCD pathogenesis. Fajgenbaum et al recently proposed a taxonomy to distinguish MCD on the basis of the HHV8 status, which provided a more accurate description of the pathogenesis and response to treatment ().Citation16
Figure 2 Proposed classification distinguishes multicentric Castleman’s disease on the basis of human herpesvirus 8 status.
Abbreviations: HHV8, human herpesvirus 8; HIV, human immunodeficiency virus; iMCD, idiopathic multicentric Castleman’s disease.
HHV8-negative MCD (idiopathic MCD [iMCD])
The presence of a subset of HHV8-negative MCD cases has long been acknowledged. Fajgenbaum et al proposed the term iMCD for HHV8-negative MCD.Citation16 Although the etiology of iMCD remains unclear, several experts suggest that proinflammatory hypercytokinemia due to viral infection, inflammation, or neoplastic disease might be a mechanism.Citation17,Citation18 Fajgenbaum et al also proposed that one or more of the following three candidate processes might be responsible for hypercytokinemia in iMCD:Citation16 autoimmune mechanisms by autoantibody-mediated antigenic stimulation or a germ-line genetic aberration in innate immune system (systemic inflammatory disease hypothesis), ectopic cytokine secretion by benign or malignant tumor cells within lymph nodes or extranodal tissue (paraneoplastic syndrome hypothesis), and viral signaling by a non-HHV8 virus (virally driven hypothesis).
Introduction of TAFRO syndrome as an iMCD subtype
Takai et al proposed a new variant of iMCD, whose features included TAFRO in their reported case series and named this variant TAFRO syndrome after its characteristic presentation.Citation8 Lymph node biopsies of these cases demonstrated HV-type-like changes consistent with MCD. These patients with TAFRO features responded well to immunosuppressive therapy with glucocorticoids and cyclosporine. They considered this novel clinical entity of iMCD to represent a group of systemic inflammatory disorders with roots in autoimmune pathophysiology, on the basis of their favorable response to immunosuppressive therapy.Citation19
Clinical features of iMCD
Liu et al published a systematic review of the literature on iMCD, providing information on its clinical features, treatment, and outcomes.Citation20 Eighty-four articles fit all the inclusion criteria, and 128 patients were included in the review. They showed that the clinical features of iMCD included systemic lymphadenopathy (128/128); anemia (79/91); elevated CRP (65/79); hypergammaglobulinemia (63/82); hypoalbuminemia (57/63); elevated IL-6 (57/63); hepatomegaly or splenomegaly (52/67); fever (33/64); edema, ascites, anasarca, or a combination of edema and anasarca (29/37); elevated soluble IL-2 receptor (20/21); and elevated VEGF (16/20). First-line treatment for iMCD included corticosteroids (47/128 [37%]), cytotoxic chemotherapy (47/128 [37%]), and anti-IL-6 therapy (11/128 [9%]). The side effect profiles revealed that anti-IL-6 therapy demonstrated better tolerability than cytotoxic agents. Anti-IL-6 might require long-term administration because relapses were reported following cessation.Citation21 The systematic review also showed that 49 of 116 patients (42%) failed first-line therapy, 2-year survival was 88% (95% confidence interval [CI] 81–95; 114 patients, 12 events, 36 events censored), and 27 of 121 (22%) patients died by the end of the observed follow-up period (median, 29 months; interquartile range, 12–50 months). Factors negatively associated with survival reportedly included age (>37 years), extravascular fluid overload, PC-type histopathology, TAFRO features, hypergammaglobulinemia, and thrombocytopenia. The 2-year survival rate was 85% (95%CI 71–100) for iMCD patients with TAFRO features versus 92% (95%CI 85–99) for those without TAFRO features (hazard ratio 2.67, 95%CI 0.4–11.20; p=0.16). The increase in mortality among patients with TAFRO syndrome within the first 6 months of diagnosis is notable because it might reflect a highly aggressive phenotype. In addition, 24 of 128 patients (19%) with iMCD had a diagnosis of malignant disease, a frequency significantly higher than that in the age-matched controls (6%). Liu et alCitation20 suggested several possible explanations for the increased prevalence of malignancies in iMCD. First, malignant cells might secrete IL-6 and other proinflammatory cytokines, leading to the histopathological and clinical features of iMCD. Second, iMCD might be a premalignant disease that eventually transforms into a malignancy. Third, an unknown genetic mutation might render the patient susceptible to both iMCD and malignant diseases. Fourth, excessive cytokine release might induce malignant transformation. Fifth, treatment modalities for iMCD such as cytotoxic therapy might amplify susceptibility to malignant disease. Finally, an unidentified virus might cause both iMCD and the malignant disease.Citation16 The authors suggested that clinicians should search for underlying malignant diseases in all patients who were newly diagnosed with iMCD and monitor those with iMCD for the development of malignant diseases.
Clinical and pathological presentation of TAFRO syndrome
Iwaki et al analyzed the clinical features and pathological characteristics of iMCD in 23 cases from Japan and two cases from the USA.Citation22 In their review, the authors categorized iMCD into two subtypes: iMCD with TAFRO symptoms (TAFRO-iMCD) and iMCD-not otherwise specified (iMCD-NOS) and found that TAFRO-iMCD was characterized by a more aggressive clinical course, refractoriness to corticosteroid therapy, more severe thrombocytopenia, higher frequency of anasarca, elevated levels of alkaline phosphatase, and almost normal gammaglobulin levels, compared with iMCD-NOS. These unique clinical and laboratory features suggested TAFRO-iMCD as a distinct variant of iMCD. This case series showed that IL-6 might not be a critical cytokine in TAFRO-iMCD, given that its levels were only modestly elevated in most cases of TAFRO-iMCD. Furthermore, the clinical features associated with elevated IL-6, such as thrombocytosis and polyclonal hypergammaglobulinemia, were not observed in TAFRO-iMCD cases. This finding contrasts with that of the control cases of iMCD-NOS. TAFRO-iMCD cases demonstrated thrombocytopenia with hyperplasia or normoplasia of megakaryocytes in the bone marrow at the time of diagnosis, possibly owing to increased peripheral thrombocyte consumption. Before the concepts of iMCD and TAFRO syndrome were established, Kojima et al classified Japanese MCD cases into two variants: idiopathic plasmacytic lymphadenopathy with polyclonal hyperimmunoglobulinemia (IPL) type and non-IPL type.Citation23 Non-IPL MCD cases showed HV-type features, proliferation of vascularity in the interfollicular zone, and a higher incidence of pleural effusion and/or ascites, thrombocytopenia, and autoimmune diseases. They also reported that non-IPL cases exhibited FDC networks with expanded or disrupted follicles, which were observed in the TAFRO-iMCD group in the study by Iwaki et al.Citation22 Conversely, IPL MCD cases exhibited clinical and pathological features more consistent with those of the iMCD-NOS group. On the basis of these findings, they speculated that TAFRO-iMCD might be immunological in origin. HHV8 status is not the sole basis of classification for MCD. Moreover, iMCD may be further categorized into iMCD with TAFRO features or iMCD-NOS as Iwaki et al demonstrated in their case series.Citation22 Their proposed diagnostic criteria for iMCD with TAFRO features () require fulfillment of the histopathological criteria, all major criteria, and one or more of the minor criteria. Diseases that had to be excluded to reach a definitive diagnosis included rheumatological diseases such as systemic lupus erythematosus; infectious diseases such as acute Epstein–Barr virus; and neoplastic diseases such as lymphoma, POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes) syndrome, and other cancers. The characteristic lymph node findings in TAFRO-iMCD included atrophic germinal centers with enlarged endothelial cell nuclei, proliferation of endothelial venules with an enlarged nucleus in the interfollicular zone, and a small number of mature PCs.
Table 1 Proposed diagnostic criteria for TAFRO-iMCD
Approach to establishing diagnostic criteria for TAFRO syndrome in Japan
Kawabata et al proposed an MCD subtype called Castleman–Kojima disease, which they defined as a systemic inflammatory disorder in patients with HHV8-negative MCD in the absence of any known autoimmune or lymphoproliferative diseases.Citation24 Castleman–Kojima disease can involve bone marrow, pleura, peritoneum, kidneys, liver, or lymph nodes. They also suggested that non-IPL MCD encompassed Castleman–Kojima disease, IgG4-related disease, POEMS syndrome, and malignant lymphomas.Citation24 shows the criteria proposed for the diagnosis of Castleman–Kojima disease, which can also be used as the diagnostic criteria for TAFRO syndrome.Citation24
Table 2 Definition of Castleman–Kojima disease (TAFRO syndrome)
Iwaki et al attempted to develop a precise definition of TAFRO symptoms in their clinicopathological review of 25 cases. In that review, thrombocytopenia was defined as a platelet count <100,000/mm3 at the time of diagnosis. Fever was defined as a temperature >38.0°C (100.4 °F). Anasarca was defined as the presence of pleural fluid and ascites by computed tomography (CT). Organomegaly included lymphadenopathy, hepatomegaly, and splenomegaly, which were confirmed by the evaluation of the CT scan by a radiologist. Reticulin fibrosis was assessed by bone marrow biopsy.Citation22
The first international consensus diagnostic criteria for iMCD
The lack of established diagnostic criteria or disease-specific biomarkers for iMCD might hinder clinicians from reaching the correct diagnosis and starting treatment before the development of organ dysfunction and death. Clinical and pathological diagnostic criteria are urgently needed to facilitate disease recognition, diagnostic evaluation, and research on pathogenesis and treatment. In 2012, Fajgenbaum et al assembled a group of physicians, researchers, and patients to create the Castleman Disease Collaborative Network (CDCN), with the aim to accelerate research through a targeted, collaborative, and patient-centric approach.Citation25 The CDCN scientific advisory board assembled clinical data for 244 iMCD patients and 88 lymph node tissue biopsies for histopathologic review, including 128 cases analyzed in a systematic review of the literature on pathology-based iMCD.Citation20 Thirty-seven cases were submitted by the CDCN working group members, and 79 cases derived from a randomized controlled study of the anti-IL-6 chimeric monoclonal antibody siltuximab in subjects with symptomatic iMCD.Citation26 An international symposium sponsored by the CDCN and the University of Pennsylvania Orphan Disease Center was held in 2015 to establish the diagnostic criteria for iMCD. All votes were anonymous, and 75% agreement was needed to pass an individual decision. The final criteria vote required 100% consensus. On the basis of the consensus at this symposium, Fajgenbaum et al recently published the multidisciplinary, evidence-based, diagnostic criteria for iMCD.Citation27 The three-part criteria were finally accepted by the working group (). A patient must meet both major criteria and fulfill at least two of the 11 minor criteria including at least one laboratory abnormality for a diagnosis of iMCD. In addition, diseases listed in the exclusion criteria must be ruled out.
Table 3 Consensus diagnostic criteria for iMCD
Treatment for iMCD and TAFRO syndrome
Corticosteroids
Corticosteroids are immunosuppressive drugs that treat both acute and chronic inflammation by decreasing transcription of proinflammatory cytokines and chemokines, adhesion molecules, and key enzymes in the inflammatory process such as IL-2, IL-6, and tumor necrosis factor-α. Corticosteroids are used as first-line treatment for iMCD including TAFRO syndrome.Citation28 However, patients frequently relapse, and corticosteroids are usually used in combination with other drugs. Nonresponders to corticosteroids are usually treated with various alternative drugs including anti-IL-6 and the chemotherapeutic agents anakinra, bortezomib, and thalidomide. However, there are limited data on prognosis and response to these treatments.
Anti-IL-6 therapies
Siltuximab
Siltuximab is a human–murine chimeric monoclonal antibody that binds to IL-6 with high affinity. A Phase III clinical trial suggested that siltuximab and optimal supportive care were superior to optimal supportive care alone and that the drug was well tolerated even during prolonged administration. Siltuximab also had a favorable effect on radiological response in lymph nodes, anemia, other disease symptoms, and inflammatory parameters. Furthermore, siltuximab therapy was not associated with a significant increase in adverse events compared with placebo.Citation26 However, siltuximab is approved for the treatment of MCD only in North America and Europe.
Tocilizumab
Tocilizumab is a humanized IL-6 antagonist capable of blocking transmembrane signaling of IL-6.Citation29 It reduces inflammation related to the IL-6 signaling cascade. Tocilizumab is currently approved for the treatment of MCD in Japan and rheumatoid arthritis worldwide.Citation30
Thalidomide
Thalidomide is an immune modulator that inhibits tumor necrosis factor-α, IL-1, IL-6, IL-12, and VEGF, while also stimulating T cells.Citation31 Thalidomide has demonstrated efficacy in inducing remission, decreasing IL-6 levels, and lowering CRP in patients with iMCD.Citation32,Citation33
Anti-CD20 therapies
Rituximab
Rituximab is a chimeric monoclonal antibody that binds to CD20, which is approved for the treatment of non-Hodgkin lymphoma. Rituximab is frequently used as first- or second-line therapy for MCD. One case report suggested that rituximab might be a therapeutic option for iMCD cases associated with immune-related disorders.Citation34 However, rituximab might be only partially effective and does not provide long-term remission if the treatment is initiated early in the course of disease before poor performance status scores decline.Citation35
Cytotoxic chemotherapy
Cytotoxic, lymphoma-based chemotherapeutic agents including cyclophosphamide, doxorubicin, vincristine, and prednisone induce responses in a large portion of the severely ill iMCD patients; however, relapses are common, and the side effects are significant.Citation36
Bortezomib
Recently, therapeutic approaches to target pathways upstream of IL-6 in iMCD have been reported, which need further evaluation, particularly for anti-IL-6 therapy-refractory patients. Bortezomib, a selective proteasome inhibitor that preferentially targets PCs, was reported to lower IL-6 levels and induce remission in four iMCD cases.Citation37,Citation38 Bortezomib might work in iMCD via direct inhibition of nuclear factor-kappa B by degrading the IkB kinase.Citation39
Future perspectives on TAFRO syndrome in iMCD
Recent case series and systematic reviews demonstrated that iMCD and TAFRO syndrome include heterogeneous diseases driven by pathological hypercytokinemia due to an unknown etiology, as previously mentioned. Although the etiology and the appropriate treatment strategies remain elusive, TAFRO syndrome might have a unique pathogenesis distinct from that of iMCD-NOS. For instance, the case series by Iwaki et al showed little evidence of an association between serum IL-6 concentration and the efficacy of anti-IL-6 therapy in TAFRO syndrome, suggesting that an elevated serum IL-6 level might not be the primary pathogenesis driving proinflammatory hypercytokinemia.Citation22 Furthermore, a clinicopathological review suggested that none of the TAFRO-iMCD cases demonstrated hypergammaglobulinemia, whereas almost all iMCD-NOS cases exhibited polyclonal hypergammaglobulinemia. Therefore, different therapeutic approaches are needed for TAFRO patients. To facilitate early diagnosis and treatment, a precise classification of the iMCD subtypes is necessary.
Recent studies have shed more light on TAFRO syndrome. First, two large systematic reviews clearly characterized the clinical manifestations and pathological findings of iMCD and TAFRO.Citation20,Citation22 Second, an international consensus on the diagnostic criteria has been established by the CDCN leadership, which further clarified the laboratory and clinical features of MCD using a global patient registry and case reports.Citation27 The etiology of iMCD awaits a similar clarification to improve disease management and treatment outcomes.
Conclusion
TAFRO syndrome is a recently established, distinct variant of HHV8-negative MCD with an autoimmune etiology. The characteristics include more aggressive clinical course, more severe thrombocytopenia, higher frequency of anasarca, and almost normal serum gammaglobulin levels compared with those of non-TAFRO iMCD. The recently established international consensus diagnostic criteria for iMCD will accordingly facilitate the identification of this syndrome. Further studies are needed to establish the clinical and pathological features of TAFRO syndrome by identifying biomarkers, pathophysiology, etiology, and treatment strategies based on evidence.
Disclosure
The authors report no conflicts of interest in this work.
References
- CastlemanBIversonLMenendezVPLocalized mediastinal lymphnode hyperplasia resembling thymomaCancer19569482283013356266
- CastlemanBTowneVWCase records of the Massachusetts General Hospital; weekly clinicopathological exercises; founded by Richard C. CabotN Engl J Med19542511039640013194083
- GabaARSteinRSSweetDLVariakojisDMulticentric giant lymph node hyperplasiaAm J Clin Pathol19786918690619617
- BowneWBLewisJJFilippaDAThe management of unicentric and multicentric Castleman’s disease: a report of 16 cases and a review of the literatureCancer199985370671710091744
- WaterstonABowerMFifty years of multicentric Castleman’s diseaseActa Oncol200443869870415764213
- KellerARHochholzerLCastlemanBHyaline-vascular and plasma-cell types of giant lymph node hyperplasia of the mediastinum and other locationsCancer19722936706834551306
- CroninDMPWarnkeRACastleman disease – an update on classification and the spectrum of associated lesionsAdv Anat Pathol200916423624619546611
- TakaiKNikkuniKShibuyaHHashidateHThrombocytopenia with mild bone marrow fibrosis accompanied by fever, pleural effusion, ascites and hepatosplenomegalyRinsho Ketsueki2010515320325 Japanese20534952
- KawabataHKadowakiNNishikoriMClinical features and treatment of multicentric Castleman’s disease: a retrospective study of 21 Japanese patients at a single instituteJ Clin Exp Hematop2013531697723801137
- ShahidiHMyersJLKvalePACastleman’s diseaseMayo Clin Proc199570109699777564550
- SoumeraiJDSohaniARAbramsonJSDiagnosis and management of Castleman diseaseCancer Control201421426627825310208
- DossierAMeigninVFieschiCBoutboulDOksenhendlerEGalicierLHuman herpesvirus 8-related Castleman disease in the absence of HIV infectionClin Infect Dis201356683384223223599
- LeroySMoshousDCassarOMulticentric Castleman disease in an HHV8-infected child born to consanguineous parents with systematic reviewPediatrics20121291e199e20322157133
- KishimotoTIL-6: from its discovery to clinical applicationsInt Immunol201022534735220410258
- StebbingJAdamsCSanittAPlasma HHV8 DNA predicts relapse in individuals with HIV-associated multicentric Castleman diseaseBlood2011118227127521511959
- FajgenbaumDCVan RheeFNabelCSHHV-8-negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapyBlood2014123192924293324622327
- SudaTKatanoHDelsolGHHV-8 infection status of AIDS-unrelated and AIDS-associated multicentric Castleman’s diseasePathol Int200151967167911696169
- DossierAMeigninVFieschiCBoutboulDOksenhendlerEGalicierLHuman herpesvirus 8-related Castleman disease in the absence of HIV infectionClin Infect Dis201356683384223223599
- TakaiKNikkuniKMomoiANagaiKIgarashiNSaekiTThrombocytopenia with reticulin fibrosis accompanied by fever, anasarca and hepatosplenomegaly: a clinical report of five casesJ Clin Exp Hematop2013531636823801136
- LiuAYNabelCSFinkelmanBSIdiopathic multicentric Castleman’s disease: a systematic literature reviewLancet Haematol201634e163e17527063975
- NishimotoNSasaiMShimaYImprovement in Castleman’s disease by humanized anti-interleukin-6 receptor antibody therapyBlood2000951566110607684
- IwakiNFajgenbaumDCNabelCSClinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman diseaseAm J Hematol201691222022626805758
- KojimaMNakamuraNTsukamotoNClinical implications of idiopathic multicentric Castleman disease among Japanese: a report of 28 casesInt J Surg Pathol200816439139818499694
- KawabataHTakaiKKojimaMCastleman-Kojima disease (TAFRO syndrome): a novel systemic inflammatory disease characterized by a constellation of symptoms, namely, thrombocytopenia, ascites (anasarca), microcytic anemia, myelofibrosis, renal dysfunction, and organomegaly: a statusJ Clin Exp Hematop2013531576123801135
- FajgenbaumDCRuthJRKelleherDRubensteinAHThe collaborative network approach: a new framework to accelerate Castleman’s disease and other rare disease researchLancet Haematol201634e150e15227063967
- Van RheeFWongRSMunshiNSiltuximab for multicentric Castleman’s disease: a randomised, double-blind, placebo-controlled trialLancet Oncol201415996697425042199
- FajgenbaumDCUldrickTSBaggAInternational, evidence-based consensus diagnostic criteria for HHV-8 – negative/idiopathic multicentric Castleman diseaseBlood2017129121646165828087540
- FrizzeraGBanksPMMassarelliGRosaiJA systemic lymphoproliferative disorder with morphologic features of Castleman’s disease. Pathological findings in 15 patientsAm J Surg Pathol1983732112316837832
- SatoKTsuchiyaMSaldanhaJBendigMMReshaping a human antibody to inhibit the interleukin 6-dependent tumor cell growthCancer Res19935348518568428365
- KishimotoTInterleukin-6: discovery of a pleiotropic cytokineArthritis Res Ther20068Suppl 2S2
- Lopez-GironaAMendyDItoTCereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomideLeukemia201226112326233522552008
- StarkeyCRJosteNELeeF-CNear-total resolution of multicentric Castleman disease by prolonged treatment with thalidomideAm J Hematol200681430330416550518
- TatekawaSUmemuraKFukuyamaRThalidomide for tocilizumab-resistant ascites with TAFRO syndromeClin Case Reports201536472478
- OcioEMSanchez-GuijoFMDiez-CampeloMEfficacy of rituximab in an aggressive form of multicentric Castleman disease associated with immune phenomenaAm J Hematol200578430230515795923
- GholamDVantelonJ-MAl-JijakliABourhisJ-HA case of multicentric Castleman’s disease associated with advanced systemic amyloidosis treated with chemotherapy and anti-CD20 monoclonal antibodyAnn Hematol2003821276676812898190
- ChronowskiGMHaCSWilderRBCabanillasFManningJCoxJDTreatment of unicentric and multicentric Castleman disease and the role of radiotherapyCancer200192367067611505414
- HessGWagnerVKreftAHeusselCPHuberCEffects of bortezomib on pro-inflammatory cytokine levels and transfusion dependency in a patient with multicentric Castleman diseaseBr J Haematol2006134554454516856889
- SobasMAVenceNAAriasJDLopezABRodriguezMFLopezJLBEfficacy of bortezomib in refractory form of multicentric Castleman disease associated to poems syndrome (MCD-POEMS variant)Ann Hematol201089221721919636554
- JuvekarAMannaSRamaswamiSBortezomib induces nuclear translocation of IB resulting in gene-specific suppression of NF-B-dependent transcription and induction of apoptosis in CTCLMol Cancer Res20119218319421224428