
Abstract
Evans syndrome (ES) is a rare and chronic autoimmune disease characterized by autoimmune hemolytic anemia and immune thrombocytopenic purpura with a positive direct anti-human globulin test. It is classified as primary and secondary, with the frequency in patients with autoimmune hemolytic anemia being 37%–73%. It predominates in children, mainly due to primary immunodeficiencies or autoimmune lymphoproliferative syndrome. ES during pregnancy is associated with high fetal morbidity, including severe hemolysis and intracranial bleeding with neurological sequelae and death. The clinical presentation can include fatigue, pallor, jaundice and mucosal bleeding, with remissions and exacerbations during the person’s lifetime, and acute manifestations as catastrophic bleeding and massive hemolysis. Recent molecular theories explaining the physiopathology of ES include deficiencies of CTLA-4, LRBA, TPP2 and a decreased CD4/CD8 ratio. As in other autoimmune cytopenias, there is no established evidence-based treatment and steroids are the first-line therapy, with intravenous immunoglobulin administered as a life-saving resource in cases of severe immune thrombocytopenic purpura manifestations. Second-line treatment for refractory ES includes rituximab, mofetil mycophenolate, cyclosporine, vincristine, azathioprine, sirolimus and thrombopoietin receptor agonists. In cases unresponsive to immunosuppressive agents, hematopoietic stem cell transplantation has been successful, although it is necessary to consider its potential serious adverse effects. In conclusion, ES is a disease with a heterogeneous course that remains challenging to patients and physicians, with prospective clinical trials needed to explore potential targeted therapy to achieve an improved long-term response or even a cure.
Introduction
Evans syndrome (ES) is an uncommon autoimmune disease that was defined by Robert Evans in 1951 when he studied the relationship between autoimmune hemolytic anemia (AIHA) and immune thrombocytopenic purpura (ITP). He described the first diagnosis criteria of ES, including the presence of anemia, reticulocytosis, increased blood bilirubin and fecal urobilinogen, no family history of hemolytic diseases, evidence of antibodies against erythrocytes at 37°C, hemolysis of transfused erythrocytes, the presence of purpura, prolonged bleeding time, bone marrow aspiration with normal or increased number of megakaryocytes and the absence of exogenous toxic agents or a baseline disease. Evans divided five groups of patients and demonstrated the presence of a serum agglutinating factor in patients with ITPCitation1 based on previously reported observations in guinea pigs and rabbits.Citation2 Some authors modified the definition of Evans as a destruction of at least two hematological blood lineages, as observed in patients with neutropenia, hemolytic anemia and thrombocytopenia.Citation3,Citation4 Importantly, AIHA is a complex syndrome with a warm, cold, mixed and biphasic thermal behavior of antibodies, but only warm ones characterize ES.
The typical course of ES is characterized by an heterogeneous chronic disease with clinical variability at onset, spontaneous remissions and exacerbations.Citation1,Citation5,Citation6 Its worldwide frequency is unknown; however, research of ES in AIHA is available in some cohorts that report an incidence of about 37%–73% in this setting.Citation6,Citation7 A higher rate in female gender has been reported in 60%–70% of ES patients.Citation5,Citation8–Citation10
Diagnosis
First, an exhaustive clinical history must be taken to determine the risk factors for developing ES, such as infections, malignancies, autoimmune diseases, recent vaccinations, drugs or a family history of immune disorders,Citation11 complemented with a thorough physical examination focused on signs of anemia or thrombocytopenia. It is important to mention that primary ES is a diagnosis of exclusion and secondary ES must be searched for to determine a baseline disease because treatment and responses considerably differ between primary and secondary ES.
Characterization of laboratory features requires a complete blood count and direct examination of peripheral blood; anemia, thrombocytopenia, reticulocytosis, poikilocytosis, mainly due to the presence of spherocytes,Citation5,Citation9 increased indirect bilirubin and lactate dehydrogenase and a positive direct anti-human globulin test (DAT) test confirming ongoing immune hemolysisCitation12 are to be expected. The main laboratory characteristics are summarized in .
Figure 1 Diagnostic approach for Evans syndrome.
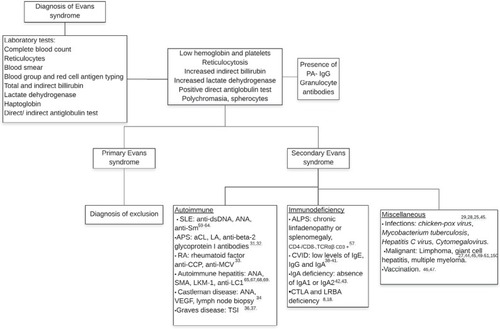
Additional studies can include the detection of antibodies against platelets (PA-IgG), documented in 35% of individuals, mostly type anti-IIb-IIIa.Citation9 Anti-granulocyte antibodies can be detected in some patients, but a lack of these antibodies does not exclude neutrophil involvement. Other antibodies must be considered to identify main autoimmune diseases associated with ES. The diagnostic work up is shown in .
Clinical features
Clinical features are associated with anemia and thrombocytopenia including pallor, weakness, fatigue, jaundice, petechiae, ecchymosis, gingivorrhagia and epistaxis.Citation5,Citation8,Citation10 Due to the prolonged immunosuppressive therapy and/or the associated underlying immune deficit, there is a risk of 66.6% of patients developing respiratory tract infections.Citation8
Differential diagnosis of ES
There are some diseases with similar characteristics to ES. Main differential diagnoses are as follows: hemolytic uremic syndrome, characterized by microangiopathic hemolytic anemia, thrombocytopenia, leukocytosis, negative DAT test, clinically manifested as diarrhea, vomiting, fever, pallor, acute renal failure in children predominantly secondary to infections with Escherichia coli, Shigella dysenteriae or Streptococcus pneumoniae;Citation13 thrombotic thrombocytopenic purpura, a disease associated with a functional deficit of ADAMTS 13 presenting as microangiopathic hemolytic anemia, thrombocytopenia, fever and neurological or kidney impairment;Citation14 Kasabach–Merritt syndrome, a rare disease predominant in early infancy with a high mortality rate of about 30% characterized by microangiopathic hemolytic anemia, thrombocytopenia, low fibrinogen levels, increment of D-dimer and giant hemangioma;Citation15,Citation16 and paroxysmal nocturnal hemoglobinuria, a chronic life-threatening disease with hemolytic anemia, pancytopenia and thrombosis of vital organs, mainly in adults. These pathologies must be excluded because treatment is considerably different in each.
Pathophysiology
Several authors have proposed different disease pathways, summarized by the presence of immune dysregulation with antibodies against erythrocytes, platelets and/or granulocytesCitation17 and decreased CD4:CD8 ratio. Mechanisms currently proposed for explaining ES are shown in ; the pathophysiology of ES remains unknown. In the following section, immunologic alterations observed in this syndrome are exposed in the larger context of autoimmunity and other relevant data are theoretically extrapolated to the disease.
Table 1 Principal mechanisms proposed to explain the pathogenesis of Evans syndrome
Deficiency of CTLA-4 (CD152) and LRBA
CTLA-4 or CD152 is an inhibitory transmembrane receptor at the surface of regulatory T-cells that bind with a high affinity to CD80/CD86 molecules in antigen-presenting cells with subsequent endocytosis and downregulation contributing to immune homeostasis. This deficiency has been related to ES;Citation18 on the other hand, LRBA is an intracellular protein that binds to CTLA-4 cytoplasmic fraction in regulatory T-cells following its endocytosis, avoiding its degradation. CTLA-4 deficiency as a cause of alteration in T-cell homeostasis has been documented in an animal model.Citation19 Recently, a gene mutation in LRBA and CTLA-4 proteins has been related to patients with ES;Citation8 also, this immune dysregulation has been found in recurrent severe opportunistic infections in humans and fatal lymphoproliferative disease in mice.Citation8,Citation20
Deficit of TPP2
A recent clinical trial revealed that tripeptidyl peptidase 2 (TPP2) is a molecule with an important role in aging, immunosenescence, autoimmunity and tumorigenesis. Its deficiency is associated with ES.Citation21 Serological analysis shows that the deficit of TPP2 is related to the presence of anti-nucleolar, anti-cytoplasmic, anti-nuclear antibodies and an increased level of age-associated B cells (ABCs), CD11c+, with a higher expression of major complex histocompatibility I molecules. This may indicate the presence of antigens on their surface, explaining the association between ABCs and autoimmune diseases.Citation21 ABCs are usually present in the elderly and have been demonstrated in patients with autoimmune diseases including systemic lupus erythematosus (SLE), rheumatoid arthritis or scleroderma and systemic sclerosis.Citation22 Further studies are needed to clearly understand the specific role of these cells in ES and autoimmune disease pathophysiology.
Decreased CD4/CD8 ratio
Abnormalities of immune regulation have been demonstrated in ES patients with a decreased level of T helper and increased T cytotoxic cells with a low CD4:CD8 ratio compared with healthy controls. Excessive production of T cytotoxic lymphocytes or a diminished activity of T helper cells could avoid the production of immunoglobulins (Ig) M and G, as observed in common variable immunodeficiency (CVID) patients.Citation17 It has been recently shown in vitro that after CD8+ T-cell activation, there are increased levels of interferon gamma with a subsequent stimulation of autoreac-tive B-cells against erythrocytes and platelets.Citation23 It could be that this increased activity of T-CD8+ cells favors deficiency of CTLA-4, which regulates inflammatory cytokine production.Citation20 These mechanisms could plausibly cause a disruption of T-cell homeostasis with an elevated concentration of interferon gamma and subsequent activation of B-cells against erythrocytes and platelets.
Classification
Classification of ES includes primary/idiopathic, with this being an exclusion diagnosis with no underlying condition, and secondary in the presence of a baseline disease. It has been demonstrated that secondary disease responds better than primary ES.Citation24 Multiple miscellaneous causes of secondary ES have been documented.Citation25–Citation52
Secondary ES
Autoimmune lymphoproliferative syndrome
Previously named Canale–Smith syndrome,Citation53 autoimmune lymphoproliferative syndrome (ALPS) is a disorder characterized by a Fas gene mutation with an alteration in T-cell apoptoic pathways causing lymphoproliferation manifested as hepatomegaly, splenomegaly, lymphadenopathies, an increased risk of Hodgkin lymphoma and immune cytopenia, mainly AIHA and ITP, with the presence of antibodies against erythrocytes and platelets,Citation54 features similar to those observed in autoimmune multilineage cytopenia.Citation10 Diagnostic criteria for ALPS were established in 1999 by the National Institutes of Health and require two necessary criteria, such as the presence of chronic lymphoproliferation, increased levels of double-negative T-cells and one primary accessory criterion with a deficit of apoptosis in vitro or genetic mutation in FAS, FASL and/or CASP 10.Citation55 Some authors suggest ruling out this diagnosis in all pediatric patients with AIHA and ES.Citation54 Others consider ALPS as a cause of secondary ES, as reported in a center with a higher frequency of concomitant ALPS and ES in 47%–50% of patients, confirmed by the presence of elevated double-negative T-cells.Citation56,Citation57 It is important to diagnose ALPS before starting treatment, since it is not recommended to administer rituximab to these patients except in specific severe unresponsive cases because of an association with developing hypogammaglobulinemia, which has been reported. CVID has also been considered as a cause of secondary ES.Citation38,Citation40 A recent study documented the increased expression of Fas in patients with ES and CVID.Citation41 Interestingly, an association between ALPS and ES has been documented only in children.Citation56,Citation57
Systemic lupus erythematosus
Several studies have demonstrated the relationship between SLE and ES;Citation9,Citation10,Citation58 prevalence of secondary ES in SLE has been reported in 1.7%–2.7% of cases.Citation24,Citation59 Hemorrhagic manifestations are frequently observed in this group, including petechiae, ecchymosis, epistaxis, gingivorraghia, gastrointestinal bleeding and hematuria.Citation5,Citation59 Spontaneous splenic rupture as an initial manifestation of ES has been documented,Citation60 confirming the heterogeneous course of the disease. Steroids are the suggested first line of therapy.Citation61 In refractory cases, rituximab at standard doses has proven effective with a reduction in PA-IgG, DAT strength,Citation62 decreased antinuclear antibodies (ANA) titer, and anti-CD19, -CD21, -B3, -TNF-α,Citation63 1–6 months after treatment.Citation62,Citation63 Autologous hematopoietic stem cell transplant in a refractory case with SLE, antiphospholipid syndrome and ES led to clinical remission, negative ANA and DAT tests, plausibly explained by a graft vs immunity effect.Citation64 Interestingly, secondary ES has a longer remission than the primary disease.Citation24
Autoimmune hepatitis
The relationship between ES and autoimmune hepatitis has been reported by few centers, with main clinical features including pallor, fatigue and splenomegaly.Citation65 Diagnosis for autoimmune hepatitis and ES is confirmed by the presence of elevated hepatic enzymes, ANA, anti-smooth muscle antibody, DAT, PA-IgG and a confirmatory biopsy with rosette formation.Citation66 Initial treatment with prednisolone and rituximab is recommended to prevent refractoriness as observed with some immunosuppressants such as azathioprine, mycophenolate mofetil (MMF) and intravenous immunoglobulin (IVIG). Improvement in cytopenias and normalization of hepatic enzymes have been observed, mainly with rituximab, in a year of follow-up.Citation65,Citation67–Citation69
Chronic lymphocytic leukemia
CLL is a worldwide common cause of hematologic cancer in the elderly.Citation70 It is frequently associated with autoimmune cytopenias.Citation70,Citation71 The frequency of ES in CLL is 2.9%.Citation72 A diagnosis of secondary ES in CLL requires the presence of AIHA and ITP at onset or within 1 month of the baseline disease. Determination of genetic mutations is necessary to establish prognosis in CLL. A 13q14 deletion is the most common chromosomic alteration and is associated with a good prognosis;Citation73 however, poor prognostic factors are predominant in patients with ES and CLL, such as a 17p13 deletion, presence of ZAP-70 and a mutation of TP53,Citation72 explaining the lower survival rate in these cases, compared to patients with only CLL.
ES during childhood
The incidence of ES in children is unknown due to a lack of published studies, with the few available consisting of case reports or retrospective cohorts.Citation6,Citation12,Citation59,Citation74–Citation76 From the largest cohorts of autoimmune cytopenias, it can be concluded that ES is more frequent during childhood; in a study, 11 (4.1%) of 179 children had ESCitation6 vs 6 (0.78%) of 766 adults.Citation77 It is important to note that autoimmune cytopenias in children are frequently associated with primary immunodeficiencies and in this group, they should always be ruled out.Citation78
A referral center of autoimmune cytopenias in France has been operating since 2004, and children with ITP, AIHA and ES have been prospectively evaluated in the OBS’CEREVANCE cohort.Citation7,Citation79 A report from this prospective cohort included 156 children from 26 different centers; median age was 5.4 years (0.2–17.2) with a male:female ratio of 1.44. Sixty-nine percent of children required more than one line of treatment, including rituximab, azathioprine, splenectomy, cyclosporine and MMF.Citation80
A large multicenter retrospective study in 21 hospitals included 42 children with a median age of 7.7 years. These patients received 1–12 modalities of treatment, mostly steroids plus IVIG, whereas others underwent splenectomy and/or received cyclosporine, vincristine, danazol, azathioprine, cyclophosphamide and plasmapheresis, with no optimal response. Twenty-four (58%) patients had complications – 12 hemorrhagic and 12 suffered severe infections, mainly sepsis, pneumonia, meningitis, abscess and osteomyelitis.Citation3
In pediatric ES cases lack of response or relapse can be severe, even after splenectomy or the use of multiple immunosuppressive agents. In addition, no large clinical trials assessing outcomes for different treatment strategies and long-term responses in children have been published.
ES during pregnancy
ES during pregnancy is not frequent and usually the diagnosis is established previously; main differential diagnoses in this circumstance are HELLP syndrome, thrombotic thrombocytopenic purpura and uremic hemolytic syndrome. There is a passive immune transfer of maternal IgG antibodies via placenta to the fetal circulation, which explains the transient thrombocytopenia or hemolytic anemia previously reported in newborns or fetus of women with autoimmune cytopenia.Citation81 In general, pregnant women with ES have a good outcome if appropriate treatment is administered opportunely; however, there is an increased risk of developing abruptio placentae and postpartum hemorrhage. Fetuses usually have a bad prognosis associated with high-risk complications occurring such as severe hemolytic anemia or hemorrhages secondary to significant thrombocytopenia, mainly intracranial bleeding with intra-extrauterine death or neurological impairments. Hematological therapy is challenging because of drugs’ teratogenic effects; treatment of choice is steroids plus IVIG. Its action mechanisms include a decreased level of antibodies crossing the placentaCitation82 and of maternal IgG antibodies by downregulation, with a reduction of these in fetal circulation.Citation83 Last treatment options are azathioprine or splenectomy, exclusively in refractory third trimester cases.Citation84
Treatment
There are no clinical trials available for ES treatment and indications for starting therapy have not been established by evidence-based studies. Few retrospective reports and small cohorts have documented responses which are summarized in .
Table 2 Second-line therapeutic options for patients with Evans syndrome
It is reasonable to treat patients with clinically significant decreased platelet and hemoglobin levels; nevertheless, as observed in ITP, the decision to start therapy in non-symptomatic patients varies in each case depending on physician experience.Citation11
The main objective consists of achieving a long-term complete response. There is no therapeutic regimen established. Steroids with and without IVIG are recommended as front-line therapy.Citation11,Citation12,Citation85 Red blood cell/platelet transfusion is indicated only in severe symptomatic patients due to the risk of exacerbations.Citation11
First-line treatments
Steroids
It is postulated that steroids inhibit the ability of macrophages for clearing platelets and erythrocytes.Citation86 Prednisolone or prednisone at 1–2 mg/kg/dayCitation11,Citation12 must be administrated in all cases, and in patients with severe clinical manifestations, an initial dose of 4–6 mg/kg/day within the first 72 hours is recommended. The initial response rate documented for patients who receive steroids at 1–2 mg/kg/day is about 82%–83%.Citation9 A regimen including a prednisolone megadose of 30 mg/kg/day for 3 days, then 20 mg/kg/day for 4 days, followed by progressive tapering to 10, 5, 2, 1 mg/kg/ week has been administered and most patients have shown complete response.Citation6,Citation11,Citation75 Three weeks after starting treatment patients need a re-evaluation; if complete response is achieved, slow tapering within 6 months is necessary to avoid adverse effects. In patients with a partial response, the initial dose of prednisolone must continue for two more consecutive weeks.Citation11 If no response is achieved, it is mandatory to start second-line therapy.Citation12 A loss of response is mainly associated with tapering the steroid dose or viral infections.Citation6 IVIG may be added if ITP is prevalent.Citation87 In cases where thrombocytopenia predominates, the presence of hepatomegaly has been related to a good response.Citation88
Intravenous immunoglobulin
Concentrated IgG from human plasma donors acts by blocking the FCγ receptor on the macrophages,Citation89 which remains a controversial therapy. In some centers, it is recommended as a first-line treatment together with steroids, and in others as a complementary drug in severe thrombocytopenic patients. A Canadian guideline recommends IVIG only in patients with predominance of ITP symptoms; in cases where AIHA symptoms are more conspicuous, other treatments are preferred.Citation90 IVIG was first indicated in 1950 for primary immune deficiencies, but it is currently an important therapy for autoimmune and hematologic diseases such as acquired hemophilia, acquired hypogammaglobulinemia, acquired von Willebrand disease, aplastic anemia, AIHA, autoimmune neutropenia and ES.Citation90 This is an important life-saving resource in uncontrolled patients;Citation11 the most common dose administered is 0.4 g/kg/day for 4 days and 5 g/kg in the case of predominance of AIHA.Citation91 Before using this therapeutic modality, it is important to consider its expense which is about 4,000 Canadian dollars per dose.Citation90 A high risk of presenting adverse effects including fever, headache, fatigue, nausea, palpitations and myalgias occurs in 20%–30% of patients.Citation92,Citation93 Severe complications have been documented, consisting mainly of acute kidney injury, arterial and venous thromboembolic events, aseptic meningitis, hemolytic anemia and infections.Citation94
Second-line treatment
Rituximab
Rituximab is a chimeric anti-CD20 targeted drug that has been increasingly used as the second-line treatment in steroid-refractory or relapsing ES. It has an immunosuppressive effect with B-cell depletion in peripheral blood after one infusion.Citation95
A pediatric multicentric prospective study analyzed the efficacy of rituximab at 375 mg/m2 in 15 patients with refractory AIHA. All patients previously received two or more courses of immunosuppressive agents (steroids, azathioprine, IVIG, cyclosporine and azathioprine) and 5/15 had ES; of these 5 patients, 3 responded with an increment in platelet levels from 27 to 140×109/L, 2 months after starting rituximab.Citation96 Another multicenter cohort retrospectively evaluated 68 adults with ES; eleven received rituximab at the standard dose with an initial response of 82% after a mean of 12 months, and two of the eleven patients relapsed.Citation9
A retrospective research investigated the use of rituximab at the standard dose plus prednisone in 17 refractory children with ES. An overall response of 76% was achieved; the main side effects during rituximab administration were vomiting, facial edema and rash, and at 1–2 weeks after infusion, transient neutropenia and pneumonia developed in one patient.Citation74 This suggested that adding rituximab to steroids can be effective in refractory cases, mainly as a second-line treatment, and emphasized the fact that repeated doses are efficient in relapsing patients. The effects of rituximab in 14 patients with immune cytopenias including 4 ES cases were reported. No patient had a concomitant response in both AIHA and ITP, suggesting that rituximab can be effective only in some patients with refractory ES.Citation97 There are ten case reports of single patients treated with rituximab, eight adults and two children; nine of these ten patients achieved remission.Citation34,Citation98–Citation106 These studies suggest that rituximab is a good option in refractory patients, and that repeated courses are necessary to achieve long-term responses. We have previously reported the outcome of six patients with ES treated at our institution, four women and two men with an age range of 10–42 years. Three received steroids as the first-line treatment and low doses of rituximab, 100 mg/week/4 weeks, as the second-line treatment with a good clinical response; after second relapse, however, two patients required splenectomy.Citation10
Mycophenolate mofetil
The 2-morpholinoethyl ester of mycophenolic acid is a selective, competitive immunosuppressant prodrug that reduces lymphocyte proliferation by inhibition of inosine monophosphate dehydrogenase.Citation107 Some authors have shown its efficacy in autoimmune cytopenias.Citation12,Citation107 A recent retrospective multicenter study evaluated the use of MMF in 40 and 16 children with ITP and ES, respectively. Most patients previously received one to three lines of treatment; 13/16 cases achieved a complete response and only 1 patient had nausea as a secondary effect.Citation108 The excellent response in that study encourages further clinical trials as it proved that MMF is safe and effective for ES, confirming its administration as a second–third line therapy.Citation11,Citation109 Effectiveness of MMF has been associated with a sustained complete remission as observed in two children; the first case was refractory to steroids and IVIGCitation110 and the second to steroids, IVIG, rituximab, vincristine and cyclophosphamide.Citation111 Both patients received MMF at 50 mg/kg/day, while the second patient also received cyclosporine A.
The use of MMF has been related to a higher risk of developing lymphoma. Nevertheless, in a prospective study of 200 patients, main adverse effects were epigastric pain and headache, with no manifestations suggestive of lymphoma.Citation112 A dose of 600 mg/m2 of MMF twice a day followed by an evaluation 3 months after the initial treatment is recommended because of the risk of relapse and scarce information on the long-term adverse effects.Citation109 These results suggest that MMF can be a useful and safe second-line treatment for ES patients refractory to steroids or IVIG.
Cyclosporine
Cyclosporine has been used in small series of patients with ES since 1994. It is an immunosuppressive drug whose mechanism of action is that it inhibits the activation of T-cells.Citation113 A total of 28 patients have been treated with this modality in different studies.Citation114–Citation120 Most were refractory to steroids, IVIG or another immunosuppressant. A response was observed in 26/28 refractory patients; thus, cyclosporine can achieve an excellent response in these circumstances. Nevertheless, it is important to carefully select patients considered for this drug in cases with no response to less-toxic drugs. If administered, continuous monitoring of serum concentrations of cyclosporine is mandatory due to the severe adverse effects associated with its administration, including malignancy and nephrotoxicity, which have been observed after administration of low doses of cyclosporine (6 mg/kg/day) after 20 years of treatment.Citation115
Splenectomy
Splenectomy was the only option in refractory relapsing patients before the introduction of rituximab, being classically considered as a second-line treatment in patients with autoimmune cytopenias, achieving a response of 60%–75% in AIHA and ITP,Citation101 while in ES, the responses are documented to be considerably heterogeneous, between 0% and 66%.Citation3,Citation6,Citation121,Citation122
Recently, splenectomy has been replaced by rituximab due to the risks of the surgical procedure. It is important to mention that in children <6 years of age, it is not a recommended option because of a higher risk of septicemia/meningitis with S. pneumoniae, Neisseria meningitidis or Haemophilus influenzae with both classic and laparoscopic techniques.Citation11,Citation12,Citation123 Also, splenectomy is contraindicated in patients with ALPS due to an elevated risk of sepsis and recurrent multilineage cytopenias.Citation124
Reported complications after splenectomy in patients with hematologic diseases include bleeding, infections and thromboembolic events.Citation125 Recently, a case report with fatal pulmonary embolism as a consequence of splenectomy in a patient with ES was published.Citation126 Laparoscopic splenectomy is an option that could avoid some secondary adverse effects, as observed in five patients with ES who underwent laparoscopic splenectomy. Two cases achieved normalization of the platelet count at a mean of 18 months and did not require additional treatment. One patient had a good initial response, but required additional medical treatment after 18 months, and two cases did not respond.Citation121
Third-line treatments
Cyclophosphamide
Cyclophosphamide is a powerful alkylating agent with anti-mitotic and immunomodulatory effects, is used in cancer and autoimmune diseases.Citation127 It is able to suppress an ongoing immune response. Although few centers have analyzed the use of cyclophosphamide in ES, a slow but progressive response has been described.Citation128 In a report, 48 patients with autoimmune cytopenias associated with CLL were evaluated. Eight had ES and were treated concomitantly with rituximab, dexamethasone and cyclophosphamide, achieving an overall response of 75%. The duration of response of CLL patients was shorter in cases of ES compared to AIHA or pure red cell aplasia.Citation129 In another center, cyclophosphamide was administered in a refractory child at a dose of 500 mg/m2 intravenously (IV), achieving a good response with improvement of hemoglobin and platelet levels.Citation130 It is known that the use of this drug is associated with a higher frequency of adverse effects, including infections and secondary malignancies.Citation128 Current results suggest that cyclophosphamide can induce remission in ES patients and it could be administered as a third–fourth-line option in refractory cases.
Alemtuzumab
A humanized monoclonal antibody against CD52 has recently been tested for AIHA and ITP and was found to show a good response.Citation131,Citation132 This antibody exerts its action through depleting lymphocytes; recently, we studied this therapeutic option in refractory ITP and AIHA at 10 mg on days 1 and 3 administered subcutaneously in combination with subcutaneous rituximab at 100 mg/week/month, and the response was 100%.Citation133 A prospective cohort of 21 patients with autoimmune cytopenias received alemtuzumab IV at 20 mg/day for 10 days; 3 patients had ES, 2 responded, whereas the third had a transient response. The first patient achieved a response within 1 week, but relapsed at 3 months and received cyclosporine and a second course of alemtuzumab. The second patient responded 3 days after administration of treatment, but relapsed at 3 months and received a second course of alemtuzumab; however, this patient died due to a cerebral hemorrhage. The last patient achieved a transient response and died because of lung cancer.Citation134
Thrombopoietin-receptor agonists
Second generation of thrombopoietin receptor agonists, romiplostim and eltrombopag, had a potential effect stimulating thrombopoiesis as observed in ITP,Citation135 and recently in ES, as documented in a patient refractory to steroids, splenectomy, IVIG and rituximab who received eltrombopag, romiplostim plus steroids with a duration of remission of 40 days at the last follow-up, suggesting that the combination of drugs is effective in decreasing platelet destruction and increasing the proliferation and maturation of megakaryocytic cells, as observed with steroids and thrombopoietin receptor agonists, respectively; this could be a highly effective double-targeted therapeutic approach.Citation136 Another center reported a patient refractory to rituximab who received romiplostim at 1–2 µg/ kg/week/4 doses and after splenectomy was performed, with a remission duration of 153 days since the first romiplostim administration.Citation137
Hematopoietic stem cell transplantation
Scarce reports have documented the use of stem cell transplantation in ES. The main benefit of allogeneic HSCT is that it currently constitutes the only curative treatment option for ES by resetting the immune system;Citation138 thus, this therapy could be a valuable alternative for patients with multiple relapses, with severe complications affecting their quality of life and/or if a lack of response to immunosuppressive drugs exists.
Allogeneic HSCT
In 1997 the first case report of a child with ES treated with ten different therapeutic immunosuppressive lines without a good response was published. The patient suffered severe bleeding (intracranial, gastrointestinal) and considerable neurological impairment. Due to his unresponsive disease, an allogeneic cord blood transplant was performed with a conditioning regimen of 225 cGy daily for 6 days plus IV cyclophosphamide, 60 mg/kg for 2 days. This patient achieved remission; nevertheless, at day 286 posttransplant, the patient developed streptococcal pharyngitis and, subsequently, liver failure, coma and died.Citation139 An adult with refractory ES treated with multiple interventions received an allogeneic HSCT with a conditioning regimen of cyclophosphamide, 200 mg/kg plus anti-thymocyte globulin at 90 mg/kg. Three months after transplant, antibodies against erythrocytes and platelets were negative with 100% chimerization. He maintained complete remission for the 30 months of follow-up.Citation140
Autologous HSCT
A prospective non-randomized study analyzed the use of cyclophosphamide at 50 mg/kg/day with autologous depleted peripheral blood stem cells for ITP in five cases with ES. They received 6–14 lines of treatment prior to transplant. Three achieved complete response and two did not respond. All cases presented bacteremia and one died after developing multiple myeloma.Citation141
In another case, a female adult received an autologous HSCT. She suffered SLE plus antiphospholipid syndrome and suddenly developed florid ES. The conditioning regimen was cyclophosphamide at 50 mg/kg/day, anti-T lymphocyte globulin 10 mg/kg/day and 6-methylprednisone 1 g/day. The patient achieved a good response with normal blood counts.Citation64
A child with refractory ES received a double autologous stem cell transplant and achieved a short-term response. On relapse, unrelated umbilical cord blood transplantation was performed with a conditioning regimen of busulfan, thiotepa, etoposide and antithymocyte globulin. After cord blood transplant, the patient achieved a good response.Citation142
The largest retrospective study of autoimmune cytopenias treated with HSCT included 36 patients, wherein 7 had ES. Five received an allogeneic HSCT and one achieved remission, two died and two were not evaluated. This suggests that auto-HSCT can be an alternative in severe autoimmune cytopenias.Citation143
Graft vs host disease has an important effect in eliminating auto-antibodies against platelets and erythrocytes, with a higher rate and prolonged remission in secondary cases of ES. The high risk of morbidity has restricted the use of HSCT for treating ES and other autoimmune diseases. Thus, results suggest that it is important to start therapy with medical treatments. If there is no response or the patient has a chronically relapsing course and if a human leukocyte antigen-identical sibling donor is available, allogeneic HSCT is preferred. In older patients with significant comorbidities who lack a human leukocyte antigen-identical donor, autologous HSCT could be performed.Citation144
ES secondary to solid organ or HSCT
Solid organ and bone marrow transplantation has been associated to the development of ES in some centers.Citation145–Citation151 The main mechanism behind this association is still unknown; immunosuppression secondary to chemotherapy of maintenance regimen could be implicated to an immune dysregulation of T-cells.Citation145 In 19 infants with genetic diseases, a mismatched cord blood transplantation was performed with a conditioning regimen of busulfan, cyclophosphamide and antithymocyte globulin; 10 cases developed chronic graft vs host disease with autoimmune cytopenias and 3 had ES.Citation146 A child with recurrent Hodgkin lymphoma underwent an autologous blood stem cell transplantation, after 15 months the patient developed ES refractory to steroids and IVIG; cyclosporine A was effective in this case.Citation147 A female patient with recurrent Hodgkin disease treated with an autologous bone marrow transplant with a conditioning regimen of cyclophosphamide, etoposide and carmustine developed ES.Citation148 A patient with non-Hodgkin lymphoma with multiple relapses received a peripheral blood stem cell transplant with a conditioning regimen of etoposide, carboplatin and cyclophosphamide; 49 days after transplantation the patient developed ES, non-Hodgkin lymphoma relapsed and died.Citation149 Also, development of refractory ES in a patient with multiple myeloma treated with an autologous stem cell transplantation has been documented.Citation150
Additional potential supporting interventions
Anecdotic reports suggest that immunological benefits can be gained in patients with autoimmune diseases including SLE and rheumatoid arthritis from simple measures, including oral honey drink and Nigella sativa oilCitation152 or powder as nutritional support measures. It is reported that wet cupping therapy (Al-Hijamah) can clear patients’ blood from autoantibodies, inflammatory mediators and immunological noxious substances.Citation153,Citation154
Perspectives in ES
New treatment approaches for patients suffering from ES should led to long-lasting remissions, less-severe relapses and an increased opportunity for cure. These scenarios will be possible as knowledge of the molecular landscape in this intriguing disease deepens, thus allowing the design of sophisticated biological therapy and/or improved HSCT strategies for severe cases, particularly in children.
A Phase II active clinical trial (Identifier: NTC00392951)Citation155 recently evaluated sirolimus at 3 mg/m2 and then at 2.5 mg/m2 depending on its serum levels in refractory autoimmune cytopenias including ES. A total of 30 patients were evaluated, including 8 with ES. There were three complete responses, three partial responses and two patients with no response. Main objectives were to document toxicities of sirolimus, including mucositis, elevation of triglycerides and cholesterol and to assess the efficacy of this approach in patients with refractory ES.Citation156
Conclusion
ES is a rare disease with a heterogeneous course characterized by multiple relapses during lifetime, despite multimodal treatment. A combination of genetic and epigenetic factors appears to be involved in its pathogenesis, and the knowledge of its precise etiology will help to design specific targeted therapies with less-severe adverse effects, significantly improving patients quality of life. Results with currently available therapeutic modalities are unsatisfactory and the disease runs a chronic relapsing course, eventually complicated by catastrophic clinical events that can lead to death. Prospective studies and clinical trials derived from knowledge on its pathophysiology are needed to improve the long-term response and even cure individuals suffering ES.
Acknowledgments
We thank Sergio Lozano-Rodriguez, MD, for his critical review of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
- EvansRTakahashiKDuaneRTPayneRLiuCPrimary thrombocytopenic purpura and acquired haemolytic anemia: evidence for a common etiologyAMA Arch Intern Med1951871486514782741
- BedsonSPBlood-Platelet anti-serum, its specificity and role in the experimental production of purpuraJ Pathol Bacteriol19222494104
- MathewPChenGWangWEvans syndrome results of a national surveyJ Pediatr Hematol Oncol19971954334379329465
- SavasanSWarrierIRavindranathYThe spectrum of Evans’ syndromeArch Dis Child19977732452489370906
- DhingraKKJainDMandalSKhuranaNSinghTGuptaNEvans syndrome: a study of six cases with review of literatureHematology200813635636019055865
- PuiCHWilimasJWangWEvans syndrome in childhoodJ Pediatr19809757547587191890
- AladjidiNLevergerGLeblancTNew insights into childhood autoimmune hemolytic anemia: a French national observational study of 265 childrenHaematologica201196565566321228033
- BesnardCLevyEAladjidiNPediatric-onset Evans syndrome: heterogeneous presentation and high frequency of monogenic disorders including LRBA and CTLA4 mutationsClin Immunol2018188525729330115
- MichelMChanetVDechartresAThe spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 casesBlood2009114153167317219638626
- Jaime-PérezJCGuerra-LealLNLópez-RazoONMéndez-RamírezNGómez-AlmaguerDExperience with Evans syndrome in an academic referral centerRev Bras Hematol Hemoter201537423023526190425
- NortonARobertsIManagement of Evans syndromeBr J Haematol2006132212513716398647
- MianoMHow I manage Evans syndrome and AIHA cases in childrenBr J Haematol2016172452453426625877
- CorriganJJJrBoineauFGHemolytic-uremic syndromePediatr Rev2001221136536911691946
- JolyBSCoppoPVeyradierAThrombotic thrombocytopenic purpuraBlood2017129212836284628416507
- KimJAChoiYBYiESExcellent outcome of medical treatment for Kasabach–Merritt syndrome: a single-center experienceBlood Res201651425626028090488
- MaguinessSGuentherLKasabach–Merritt syndromeJ Cutan Med Surg20026433533911951132
- WangWHerrodHPuiCHPresburyGWilimasJImmunoregulatory abnormalities in Evans syndromeAm J Hematol19831543813906606357
- SchmidtREGrimbacherBWitteTAutoimmunity and primary immunodeficiency: two sides of the same coin?Nat Rev Rheumatol2018141718
- LoBFritzJMSuHCCHAI and LATAIE: new genetic diseases of CTLA-4 checkpoint insufficiencyBlood201612881037104227418640
- FriedlineRHBrownDSNguyenHCD4+ regulatory T cells require CTLA-4 for the maintenance of systemic toleranceJ Exp Med2009206242143419188497
- StepenskyPRensing-EhlAGatherREarly-onset Evans syndrome, immunodeficiency, and premature immunosenescence associated with tripeptidyl-peptidase II deficiencyBlood2015125575376125414442
- RubtsovAVRubtsovaKFischerAToll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c+ B-cell population is important for the development of autoimmunityBlood201111851305131521543762
- KarakantzaMMouzakiATheodoropoulouMBusselJBManiatisATh1 and Th2 cytokines in a patient with Evans’ syndrome and profound lymphopeniaBr J Haematol2000110496897011054090
- CostallatGLAppenzellerSCostallatLTEvans syndrome and systemic lupus erythematosus: clinical presentation and outcomeJoint Bone Spine201279436236421944976
- LambotteOGelu-SimeonMMaigneGPegylated interferon alpha-2a-associated life-threatening Evans’ syndrome in a patient with chronic hepatitis CJ Infect2005513e113e11516230187
- ShivalingaiahSKArpanaJJainMKVargheseVKHyperhomocysteinemia and Evan′s syndrome with uncal herniation for emergency splenectomyAnesth Essays Res20159111812025886436
- YokoyamaSKasaharaMFukudaAEvans syndrome after successful living-donor liver transplantation for neonatal giant cell hepatitisTransplantation200784679879917893616
- SharmaSDasguptaSSumanSKKumarUChitekelaSDisseminated tuberculosis with Evans syndrome: an uncommon presentationTrop Doct201747217918128118797
- TanakaYMasuyaMKatayamaNDevelopment of mixed-type autoimmune hemolytic anemia and Evans’ syndrome following chicken pox infection in a case of low-titer cold agglutinin diseaseInt J Hematol200684322022317050195
- ShatzelJJDonohoeKChuNQProfound autoimmune hemolysis and Evans syndrome in two asplenic patients with babesiosisTransfusion201555366166525354478
- FontJJiménezSCerveraRSplenectomy for refractory Evans’ syndrome associated with antiphospholipid antibodies: report of two casesAnn Rheum Dis2000591192092311053074
- RückertAGlimmHLübbertMGrüllichCSuccessful treatment of life-threatening Evans syndrome due to antiphospholipid antibody syndrome by rituximab-based regimen: a case with long-term followupLupus200817875776018625656
- MassonCBrégeonCIfrahNBertonVHousseauFRénierJCEvans’ syndrome caused by D-penicillamine in rheumatoid arthritis. Value of the corticoids–danazol combinationRev Rhum Mal Osteoartic1991587519522 French1925397
- QuinnJPGilliganOMHorganMEvan’s syndrome complicating multicentric Castleman’s disease – dramatic response to rituximabEur J Haematol200473538438515458520
- DavePKrishnaKDiwanAGEvan’s syndrome revisitedJ Assoc Physicians India2012606061
- IkedaKMaruyamaYYokoyamaMAssociation of Graves’ disease with Evans’ syndrome in a patient with IgA nephropathyIntern Med200140101004101011688823
- YashiroMNagoshiHKasugaYEvans’ syndrome associated with Graves’ diseaseIntern Med199635129879909031001
- García-MuñozRAntonJRodriguez-OteroPCommon variable immunodeficiency and Evans syndrome complicated by autoimmune hemolysis due to anti-Jka auto-antibodiesLeuk Lymphoma20084961220122218452064
- AziziGAbolhassaniHAsgardoonMHAutoimmunity in common variable immunodeficiency: epidemiology, pathophysiology and managementExpert Rev Clin Immunol201713210111527636680
- PodjasekJCAbrahamRSAutoimmune cytopenias in common variable immunodeficiencyFront Immunol2012318922837758
- Savas¸anSWarrierIBuckSKaplanJRavindranathYIncreased lymphocyte Fas expression and high incidence of common variable immunodeficiency disorder in childhood Evans’ syndromeClin Immunol2007125322422917936685
- PaaskeHSrensenCWAstrupLEvans’ syndrome in IgA deficiency. Episodic autoimmune haemolytic anaemia and thrombocytopenia during a 10 years observation periodScand J Haematol19822652707146824
- RocaBFerránGSimónECortésVLymphoid hyperplasia of the lung and Evans’ syndrome in IgA deficiencyAm J Med1999106112112210320128
- BechirAHaifaRNesrineBSMultiple myeloma associated with an Evan’s syndromePan Afr Med J20162512728292089
- YamamotoGHosoiMMiyagawaTEvans syndrome with cytomegalovirus infection followed by emerging peripheral T-cell lymphomaAnn Hematol201291112312421437588
- ShlamovitzGZJoharSA case of Evans’ syndrome following influenza vaccineJ Emerg Med2013442e149e15122796025
- MartínezEDomingoPEvans’s syndrome triggered by recombinant hepatitis B vaccineClin Infect Dis199215610511457642
- HyunDKimDJungJSohnSLeeJLeeKA case of Epstein– Barr virus infection presented as Evans syndromeBlood Res1998333438442
- OzgönenelBMoonkaDSavas¸anSFulminant hepatitis B following rituximab therapy in a patient with Evans syndrome and large B-cell lymphomaAm J Hematol2006814302
- KubotaTDaibataMKobayashiMTaguchiH[Malignant lymphoma of the lung associated with Evans’ syndrome: report of a case]. [Article in Japanese]Nihon Kokyuki Gakkai Zasshi2002401296596912692949
- D’AmbrosioDTomaselliVGargiuloGRoselliMdella-MorteDAbetePEvans syndrome presented with marginal zone lymphoma and duodenal neuroendocrine tumor in an elderly womanInt J Gerontol2016104237239
- AmidALeungEEvans syndrome secondary to HIV infectionJ Pediatr Hematol Oncol201335649049123887732
- CanaleVCSmithCHChronic lymphadenopathy simulating malignant lymphomaJ Pediatr19677068918994165068
- Garrido ColinoCAvances en el conocimiento y manejo del síndrome linfoproliferativo autoinmuneAn Pediatr2014802122.e112122
- OliveiraJBBleesingJJDianzaniURevised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International WorkshopBlood201011614e35e4020538792
- SeifAEMannoCSSheenCGruppSATeacheyDTIdentifying autoimmune lymphoproliferative syndrome in children with Evans syndrome: a multi-institutional studyBlood2010115112142214520068224
- TeacheyDTMannoCSAxsomKMUnmasking Evans syndrome: T-cell phenotype and apoptotic response reveal autoimmune lymphoproliferative syndrome (ALPS)Blood200510562443244815542578
- ThakurNChandraJDhingraBSinghVPediatric lupus: varied haematological picture and presentationIndian J Hematol Blood Transfus2015311687025548448
- LubeGEFerrianiMPCamposLMEvans syndrome at childhood-onset systemic lupus erythematosus diagnosis: a large multicenter studyPediatr Blood Cancer20166371238124327018636
- TokgozHCaliskanUAtasBOzbekOTavilBSpontaneous rupture of the spleen in a patient with systemic lupus erythematosus initially presented as Evans syndromeJ Pediatr Hematol Oncol2014361e39e4123389505
- MendoncaSSrivastavaSKapoorRGuptaDGuptaPSharmaMLEvans syndrome and its link with systemic lupus erythematosusSaudi J Kidney Dis Transpl201627114714926787583
- KittakaKDobashiHBabaNA case of Evans syndrome combined with systemic lupus erythematosus successfully treated with rituximabScand J Rheumatol200837539039318609263
- TamimotoYHoriuchiTTsukamotoHA dose-escalation study of rituximab for treatment of systemic lupus erythematosus and Evans’ syndrome: immunological analysis of B cells, T cells and cytokinesRheumatology (Oxford)200847682182718397955
- MussoMPorrettoFCrescimannoAAutologous peripheral blood stem and progenitor (CD34+) cell transplantation for systemic lupus erythematosus complicated by Evans syndromeLupus1998774924949796853
- OhkawaraHFurukawaMIkedaKSteroid-resistant autoimmune myelofibrosis in a patient with autoimmune hepatitis and Evans syndrome complicated with increased expression of TGF-β in the bone marrow: a case reportInt J Hematol2017106571872428584963
- TiniakosDGBrainJGBuryYARole of histopathology in autoimmune hepatitisDig Dis2015332536426642062
- KorkmazHBugdaciMSTemelTDagliMKarabagliPAutoimmune hepatitis–primary biliary cirrhosis overlap syndrome concomitant with immune hemolytic anemia and immune thrombocytopenic purpura (Evans syndrome)Clin Res Hepatol Gastroenterol2013372e45e5023273499
- JarasvaraparnCImranHSiddiquiAWilsonFGremseDAAssociation of autoimmune hepatitis type 1 in a child with Evans syndromeWorld J Hepatol20179231008101228878866
- CareyEJSomaratneKRakelaJSuccessful rituximab therapy in refractory autoimmune hepatitis and Evans syndromeRev Med Chil2011139111484148722446656
- O’ReillyAMurphyJRaweSGarveyMChronic lymphocytic leukemia: a review of front-line treatment options, with a focus on elderly CLL patientsClin Lymphoma Myeloma Leuk201818424925629477297
- MorenoCHodgsonKFerrerGAutoimmune cytopenia in chronic lymphocytic leukemia: prevalence, clinical associations, and prognostic significanceBlood2010116234771477620736453
- CarliGViscoCFalisiEEvans syndrome secondary to chronic lymphocytic leukaemia: presentation, treatment, and outcomeAnn Hematol201695686387027001309
- Dal BoMRossiFMRossiD13q14 Deletion size and number of deleted cells both influence prognosis in chronic lymphocytic leukemiaGenes Chromosomes Cancer201150863364321563234
- Bader-MeunierBAladjidiNBellmannFRituximab therapy for childhood Evans syndromeHaematologica200792121691169418055994
- WangWCEvans syndrome in childhood: pathophysiology, clinical course, and treatmentAm J Pediatr Hematol Oncol19881043303383071168
- DemanelisKSriplungHMezaRDifferences in childhood leukemia incidence and survival between Southern Thailand and the United States: a population-based analysisPediatr Blood Cancer201562101790179825962869
- SilversteinMNHeckFJAcquired hemolytic anemia and associated thrombocytopenic purpura: with special reference to Evans syndromeProc Staff Meet Mayo Clin19622837122128
- SeidelMGAutoimmune and other cytopenias in primary immunodeficiencies: pathomechanisms, novel differential diagnoses, and treatmentBlood2014124152337234425163701
- PasquetMAladjidiNGuitonCRomiplostim in children with chronic immune thrombocytopenia (ITP): the French ExperienceBr J Haematol2014164226627124152194
- AladjidiNFernandesHLeblancTEvans syndrome in children: long-term outcome in a prospective French national observational cohortFront Pediatr201537926484337
- SankaranSRobinsonSEImmune thrombocytopenia and pregnancyObstet Med20114414014627579112
- PhupongVSareepapongWWitoonpanichPEvans syndrome and pregnancy: a case reportBJOG2004111327427614961891
- NiHChenPSpringCMA novel murine model of fetal and neonatal alloimmune thrombocytopenia: response to intravenous IgG therapyBlood200610772976298316317099
- LefkouENelson-PiercyCHuntBJEvans’ syndrome in pregnancy: a systematic literature review and two new casesEur J Obstet Gynecol Reprod Biol20101491101720031296
- MantadakisEFarmakiENatural history, pathogenesis, and treatment of Evans syndrome in childrenJ Pediatr Hematol Oncol201739641341928654461
- MizutaniHFurubayashiTImaiYMechanisms of corticosteroid action in immune thrombocytopenic purpura (ITP): experimental studies using ITP-prone mice, (NZW × BXSB) F1Blood19927949429471737103
- Flores-MontesOAEscobar-OrduñoMCLozano-GarcidueñasMValle-LealJGSíndrome de Evans en lactantesBol Med Hosp Infant Mex201774214114629382497
- FloresGCunningham-RundlesCNewlandACBusselJBEfficacy of intravenous immunoglobulin in the treatment of autoimmune hemolytic anemia: results in 73 patientsAm J Hematol19934442372428237993
- GodeauBBierlingPTreatment of idiopathic thrombocytopenic purpura in adultsPresse Med20083791292129818644317
- AndersonDAliKBlanchetteVGuidelines on the use of intravenous immune globulin for hematologic conditionsTransfus Med Rev2007212 Suppl 1S9S5617397769
- HilgartnerMWBusselJUse of intravenous gamma globulin for the treatment of autoimmune neutropenia of childhood and autoimmune hemolytic anemiaAm J Med1987834A2529
- KatzUAchironAShererYShoenfeldYSafety of intravenous immunoglobulin (IVIG) therapyAutoimmun Rev20076425725917317619
- ShererYLevyYLangevitzPRauovaLFabrizziFShoenfeldYAdverse effects of intravenous immunoglobulin therapyPharmacology200062133137
- WangJMcquiltenZKWoodEMAubronCIntravenous immunoglobulin in critically ill adults: when and what is the evidence?J Crit Care2015303652.e9e16
- ReffMECarnerKChambersKSDepletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20Blood19948324354457506951
- ZeccaMNobiliBRamenghiURituximab for the treatment of refractory autoimmune hemolytic anemia in childrenBlood2003101103857386112531800
- ShanafeltTDMaduemeHLWolfRCTefferiARituximab for immune cytopenia in adults: idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, and Evans syndromeMayo Clin Proc200378111340134614601692
- RodellaEPacquolaEBianchiniERamazzinaEPaoliniRConsolidation treatment with rituximab induces complete and persistent remission of mixed type Evans syndromeBlood Coagul Fibrinolysis200819431531818469554
- JubinskyPTRashidNSuccessful treatment of a patient with mixed warm and cold antibody mediated Evans syndrome and glucose intolerancePediatr Blood Cancer200545334735015704210
- ParkCYChungCHA patient with mixed type evans syndrome: efficacy of rituximab treatmentJ Korean Med Sci20062161115111617179698
- MantadakisEDanilatouVStiakakiEKalmantiMRituximab for refractory Evans syndrome and other immune-mediated hematologic diseasesAm J Hematol200477330331015495242
- KnechtHBaumbergerMTobònASteckASustained remission of CIDP associated with Evans syndromeNeurology200463473073215326255
- UrbanCBeneschMSovinzPSchwingerWLacknerHFatal Evans’ syndrome after matched unrelated donor transplantation for hyper-IgM syndromeEur J Haematol200472644444715128425
- GalorAO’BrienTRituximab treatment for relapsed autoimmune hemolytic anemia in Evans syndromeInt J Hematol200378433533614686491
- SeipeltGBöhmeAKoschmiederSHoelzerDEffective treatment with rituximab in a patient with refractory prolymphocytoid transformed B-chronic lymphocytic leukemia and Evans syndromeAnn Hematol200180317017311320903
- Abdel-RaheemMMPottiAKobrinskyNSevere Evans’s syndrome secondary to interleukin-2 therapy: treatment with chimeric monoclonal anti-CD20 antibodyAnn Hematol200180954354511669305
- HowardJHoffbrandAVPrenticeHGMehtaAMycophenolate mofetil for the treatment of refractory auto-immune haemolytic anaemia and auto-immune thrombocytopenia purpuraBr J Haematol2002117371271512028047
- MianoMRamenghiURussoGMycophenolate mofetil for the treatment of children with immune thrombocytopenia and Evans syndrome. A retrospective data review from the Italian Association of Paediatric Haematology/OncologyBr J Haematol2016175349049527447678
- MianoMScalzoneMPerriKMycophenolate mofetil and Sirolimus as second or further line treatment in children with chronic refractory primitive or secondary autoimmune cytopenias: a single centre experienceBr J Haematol2015171224725326058843
- Guirat-DhouibNMellouliFKoukiRBejaouiMSuccessful treatment of mycophenolate mofetil in a child with refractory Evans syndromeJ Pediatr Hematol Oncol2010326e244
- FarruggiaPMacalusoATropiaSEffectiveness of cyclosporine and mycophenolate mofetil in a child with refractory Evans syndromePediatr Rep201132e15
- RaoVKDuganFDaleJKUse of mycophenolate mofetil for chronic, refractory immune cytopenias in children with autoimmune lymphoproliferative syndromeBr J Haematol2005129453453815877736
- MatsudaSKoyasuSMechanisms of action of cyclosporineImmunopharmacology2000472-311912510878286
- EarnshawIThachilJExample of the drug interaction between ciclosporin and orlistat, resulting in relapse of Evan’s syndromeBMJ Case Rep20162016bcr2016217246
- JanićDKrivokapiić-DokmanovićLJovanovićNLazićJRodićPJankovićSGlucocorticoid-resistant Evans’ syndrome successfully controlled with low-dose cyclosporineInt J Clin Pharmacol Ther2011491062262521961487
- UçarBAkgünNAydoğduSDKirelBIdemSTreatment of refractory Evans’ syndrome with cyclosporine and prednisonePediatr Int199941110410710200147
- RackoffWRMannoCSTreatment of refractory Evans syndrome with alternate-day cyclosporine and prednisoneAm J Pediatr Hematol Oncol19941621561598166369
- EmiliaGMessoraCLongoGBertesiMLong-term salvage treatment by cyclosporin in refractory autoimmune haematological disordersBr J Haematol19969323413448639426
- LiuHShaoZJingLThe effectiveness of cyclosporin A in the treatment of autoimmune hemolytic anemia and Evans syndromeZhonghua Xue Ye Xue Za Zhi20012211581583 Chinese11855146
- ScaradavouABusselJEvans syndrome. Results of a pilot study utilizing a multiagent treatment protocolJ Pediatr Hematol Oncol19951742902957583383
- DuperierTFelsherJBrodyFLaparoscopic splenectomy for Evans syndromeSurg Laparosc Endosc Percutan Tech2003131454712598759
- LiYPankajPWangXWangYPengBSplenectomy in the management of Evans syndrome in adults: long-term follow up of 32 patientsSurg Pract20141811522
- LeoneGPizzigalloEBacterial infections following splenectomy for malignant and nonmalignant hematologic diseasesMediterr J Hematol Infect Dis201571e201505726543526
- PriceSShawPKirkJCauses and consequences of splenectomy in ALPS-FASBlood2010116213908
- BickenbachKAGonenMLabowDMIndications for and efficacy of splenectomy for haematological disordersBr J Surg2013100679480023436638
- MongaVMaliskeSMPerepuUFatal pulmonary embolism following splenectomy in a patient with Evan’s syndrome: case report and review of the literatureThromb J2017151828680366
- AhlmannMHempelGThe effect of cyclophosphamide on the immune system: implications for clinical cancer therapyCancer Chemother Pharmacol201678466167127646791
- BrodskyRAHigh-dose chemotherapy in patients with severe autoimmune diseaseAnn Intern Med1998129103110359867758
- MichalletASRossignolJCazinBYsebaertLRituximab–cyclophosphamide–dexamethasone combination in management of autoimmune cytopenias associated with chronic lymphocytic leukemiaLeuk Lymphoma20115271401140321699387
- GombakisNTrahanaMAthanassiouMKanakoudi-TsakalidouFEvans syndrome: successful management with multi-agent treatment including intermediate-dose intravenous cyclophosphamideJ Pediatr Hematol Oncol199921324824910363861
- OsterborgAKarlssonCLundinJAlemtuzumab to treat refractory autoimmune hemolytic anemia or thrombocytopenia in chronic lymphocytic leukemiaCurr Hematol Malig Rep200941475320425438
- CheungWWHwangGYTseEKwongYLAlemtuzumab induced complete remission of autoimmune hemolytic anemia refractory to corticosteroids, splenectomy and rituximabHaematologica2006915 SupplECR1316709521
- Gómez-AlmaguerDSolano-GenestaMTarín-ArzagaLLow-dose rituximab and alemtuzumab combination therapy for patients with steroid-refractory autoimmune cytopeniasBlood2010116234783478520841509
- WillisFMarshJCBevanDHThe effect of treatment with Campath-1H in patients with autoimmune cytopeniasBr J Haematol2001114489189811564082
- Gómez-AlmaguerDColunga-PedrazaPRGómez-de LeónAGutiérrez-AguirreCHCantú-RodríguezOGJaime-PérezJCEltrombopag, low-dose rituximab, and dexamethasone combination as frontline treatment of newly diagnosed immune thrombocytopaeniaBr J Haematol Epub20171221
- Ruiz-ArguellesGJRuiz-DelgadoGJVelázquez-Sánchez-de-CimaSZamora-OrtizGSimultaneous romiplostin, eltrombopag, and prednisone were successful in severe thrombocytopenia of Evans syndrome refractory to hydrocortisone, splenectomy, intravenous IgG, and rituximabHematology201318317517723321502
- Gonzalez-NietoJAMartin-SuarezIQuattrinoSThe efficacy of romiplostim in the treatment of severe thrombocytopenia associated to Evans syndrome refractory to rituximabLupus201120121321132321719526
- VaughnJEAnwerFDeegHJTreatment of refractory ITP and Evans syndrome by haematopoietic cell transplantation: is it indicated, and for whom?Vox Sang2016110151126178735
- RaetzEBeattyPGAdamsRHTreatment of severe Evans syndrome with an allogeneic cord blood transplantBone Marrow Transplant19972054274299339762
- OyamaYPapadopoulosEBMirandaMTraynorAEBurtRKAllogeneic stem cell transplantation for Evans syndromeBone Marrow Transplant200128990390511781654
- HuhnRDFogartyPFNakamuraRHigh-dose cyclophosphamide with autologous lymphocyte-depleted peripheral blood stem cell (PBSC) support for treatment of refractory chronic autoimmune thrombocytopeniaBlood20031011717712393623
- UrbanCLacknerHSovinzPSuccessful unrelated cord blood transplantation in a 7-year-old boy with Evans syndrome refractory to immunosuppression and double autologous stem cell transplantationEur J Haematol200676652653016529601
- PasswegJRHaematopoietic stem cell transplantation for immune thrombopenia and other refractory autoimmune cytopeniasBest Pract Res Clin Haematol200417230531515302342
- BeneschMUrbanCPlatzbeckerUPasswegJStem cell transplantation for patients with Evans syndromeExpert Rev Clin Immunol20095334134820477011
- AuWYLoCMHawkinsBRMaESLieAKKwongYLEvans’ syndrome complicating chronic graft versus host disease after cadaveric liver transplantationTransplantation200172352752811502987
- PageKMMendizabalAMPrasadVKPosttransplant autoimmune hemolytic anemia and other autoimmune cytopenias are increased in very young infants undergoing unrelated donor umbilical cord blood transplantationBiol Blood Marrow Transplant200814101108111718804040
- KatoIOkadaHNishidaTEvans syndrome after autologous peripheral blood stem cell transplantation for recurrent Hodgkin lymphomaPediatr Int201658993393627577012
- KeungY-KCobosEBolanos-MeadeJIssarachaiSBrideauAMorganDEvans syndrome after autologous bone marrow transplant for recurrent Hodgkin’s diseaseBone Marrow Transplant19972012109911019466285
- KamezakiKFukudaTMakinoSHaradaMEvans’ syndrome following autologous peripheral blood stem cell transplantation for non-Hodgkin’s lymphomaClin Lab Haematol200426429129315279667
- ChiharaDSakamotoTArimoto-MiyamotoKRefractory Evans syndrome after autologous stem cell transplantation for multiple myeloma: management with a second transplantationIntern Med201049768368720371959
- Hwang-BoSKimSKLeeJWTreatment and response of autoimmune cytopenia occurring after allogeneic hematopoietic cell transplantation in childrenBlood Res201752211912428698848
- KheirouriSHadiVAlizadehMImmunomodulatory effect of Nigella sativa oil on T lymphocytes in patients with rheumatoid arthritisImmunol Invest201645427128327100726
- BaghdadiHAbdel-AzizNAhmedNSAmeliorating role exerted by Al-Hijamah in autoimmune diseases: effect on serum autoantibodies and inflammatory mediatorsInt J Health Sci (Qassim)20159220723226309442
- El SayedSMBaghdadiHAbou-TalebAAl-hijamah and oral honey for treating thalassemia, conditions of iron overload, and hyperferremia: toward improving the therapeutic outcomesJ Blood Med2014521923725382989
- TeacheyDTdatabase on the InternetChildren’s Hospital of Philadelphia. Sirolimus for autoimmune disease of blood cellsClin trials Available from: https://clinicaltrials.gov/ct2/show/NCT00392951. NLM identifier: NCT00392951Accessed May 21, 2018
- BrideKLVincentTSmith-WhitleyKSirolimus is effective in relapsed/refractory autoimmune cytopenias: results of a prospective multi-institutional trialBlood20161271172826504182
- KashifMQureshiAAdilSNKhurshidMSuccessful use of rituximab in Evans syndrome and refractory immune thrombocytopenic purpuraJ Pak Med Assoc2010601646520055286
- KotbRPinganaudCTrichetCEfficacy of mycophenolate mofetil in adult refractory auto-immune cytopenias: a single center preliminary studyEur J Haematol2005751606415946312
- BrodskyRAPetriMSmithBDImmunoablative high-dose cyclophosphamide without stem-cell rescue for refractory, severe autoimmune diseaseAnn Intern Med199812912103110359867758