
Abstract
Post transfusion purpura (PTP) is an uncommonly reported post transfusion adverse event that can present with severe thrombocytopenia; sometimes resulting in significant bleeding and hemorrhage. Its diagnosis can be elusive given its substantial symptomatic overlap with other thrombocytopenic syndromes. Underdiagnosis and underreporting make the true incidence of disease difficult to define. While clinical suspicion is key, laboratory evidence of platelet-targeted antibodies and identification of the antigen(s) they recognize are necessary to confirm the diagnosis. A curious aspect of PTP is paradoxical destruction of both transfused and autologous platelets. Although the first case was reported over 50 years ago, this aspect of PTP pathogenesis is still not fully understood and is widely debated. Several theories exist, but conclusive evidence to support most is lacking. Despite limited understanding of disease incidence and etiology, treatment with IVIG (Intravenous Immunoglobulin) has become standard practice and can be highly effective. Although recurrence is rare, precautions should be taken if patients with a history of PTP require transfusions in the future.
Introduction
Post Transfusion Purpura (PTP) is an uncommon but serious transfusion-associated complication characterized by profound thrombocytopenia developing within 2 weeks of transfusion. Bleeding of variable severity is often present and can be life-threatening. When compared to the rate of other transfusion reactions such as delayed hemolytic (1:2500–11,000), alloimmunization (1:100), urticarial (1–3%) and febrile nonhemolytic (0.1% to 1%),Citation1 PTP is considered an exceedingly rare event by many clinicians. However, the prevalence of PTP is largely unknown. Studies have reported variable incidence ranging from 1:24,000 to 1:50,000–100,000 transfusions.Citation2,Citation3 The true incidence is difficult to discern as the disease is difficult to differentiate from other thrombocytopenic conditions, and is likely under recognized and underreported.
PTP was first reported in 1961 in two multiparous women undergoing surgery.Citation4 Various case reports and series have identified previously pregnant females as the most commonly affected group although recent data suggest that this risk may decrease with advancing age in elderly females.Citation5 The likelihood of developing PTP appears to be increased by prior exposure to a specific human platelet antigen (HPA) absent on the patient’s platelets. Subsequent transfusion re-exposes the recipient to this antigen, which triggers an anamnestic alloimmune response which somehow induces autologous platelet destruction. The most commonly implicated antibody is anti-HPA-1a made by HPA-1b/1b recipients, an uncommon genotype found in ~2% of the Caucasian population.
Despite knowledge of its existence for more than 5 decades and >200 reported cases, PTP remains an elusive entity. The goal of this review is to provide a comprehensive overview of current perspectives regarding PTP including diagnosis, treatment and pathophysiology and patient outcomes.
Clinical Presentation
PTP presents as severe thrombocytopenia (<10,000 platelets/µL) sometimes heralded by life-threatening bleeding. Typically, symptoms occur 5–10 days after transfusion of platelet-containing blood products. Case series identify mucous membranes, gastrointestinal tract and urinary tract as common sites of bleeding.Citation2,Citation5 Intracranial hemorrhage and death has occurred in severe cases. Accompanying fevers, chills and platelet transfusion refractoriness have also been seen.Citation6 The condition is female predominant; most commonly identified in middle-aged multiparous women. A recent study suggests increased risk in elderly populations with underlying illnesses such as coagulopathy, cardiac arrhythmias, leukemia and transplant. The number of units transfused to a patient has also shown to have an impact on PTP occurrence.Citation5 A fatality rate ranging from 10–20% has been reported in the literature, most often related to intracranial hemorrhage.Citation1,Citation2 PTP is considered a self-limited disease with recovery of platelet counts in approximately 20 days.Citation7 The disease is mediated by an anamnestic antibody response against a transfused HPA that the patient lacks. Individuals with HPA-1b/1b genotype previously immunized via pregnancy or transfusion are the prototypical subjects of most case reports. Cases in men, albeit rare, have also been reported.Citation3,Citation8–Citation10 Alloantibodies against a common HPA antigen found on the majority of normal donor platelets are thought to be somehow responsible for destruction of platelets of the transfusion recipient even though they lack the alloantigen toward which antibody is directed.
Pathogenic Mechanism
As noted, the biological mechanism responsible for the development of PTP appears to be somehow connected to the robust, platelet-specific immune response that develops 5–10 days following blood product transfusion. Patients previously sensitized to platelet antigens through pregnancy or transfusion are re-sensitized to the same antigen(s), and produce potent platelet-reactive antibodies. As mentioned, antibodies against HPA-1a are the most frequently reported.
HPA-1a (Zw, PlA1) was the first platelet antigen to be defined. Interestingly, sera from three PTP patients was used in serologic studies that characterized the antigen. HPA-1a antibodies were first described in a 1959 report by Van Loghem et al in a female patient who developed severe thrombocytopenia 7 days after receiving a single blood transfusion.Citation11 A strong platelet antibody was detected in her serum that agglutinated platelet suspensions from 97.6% of unrelated donors and the antigen it recognized was dubbed Zw. Two years later, Shulman and colleagues described two post-surgery patients, both multiparous females, that experienced acute severe thrombocytopenia within a week of receiving blood transfusions.Citation4 Sera from both women contained strong complement fixing, platelet-agglutinating antibodies they named anti-PlA1. Sera were sent to the Van Loghem lab and testing confirmed that PlA1 and Zw antibodies recognized the same platelet antigen, now called HPA-1a. However, the conundrum of a PTP mechanism involving platelet destruction by potent alloantibodies has always been—how can HPA-1a antibodies clear both transfused HP-1a+ platelets and the patient’s autologous platelets, which do not express HPA-1a? Efforts to further elucidate the mechanism have focused on answering this question.
Three theories of autologous platelet destruction in PTP have predominated. Interestingly, all three were either first proposed or alluded to in the 1961 report of Shulman et al, in which they also coined the term “post-transfusion purpura”. The first two are both predicated on the presence of soluble HPA-1a in the transfused donor blood. One suggests that immune complexes made up of anti-HPA-1a and transfused soluble HPA-1a bind non-specifically to autologous platelets, leading to platelet clearance via macrophages in the reticuloendothelial system (RES). The second proposes that transfused soluble HPA-1a coats the patient’s own platelets making them capable of binding to and being cleared by the potent HPA-1a antibodies present. The third theory holds that platelet autoantibodies are produced concomitantly with HPA-1a alloantibodies and that the autoantibodies, not the HPA-1a antibodies, cause autologous platelet destruction. Shulman et alCitation4 primarily based their immune complex theory on similarities they recognized between PTP and their studies of drug-induced thrombocytopenia; which at the time was thought to be caused by immune complexes of drug and drug-reactive antibodies.Citation12 PTP case reports by other groups claimed binding of HPA-1a antibody/antigen immune complexes to HPA-1b/1b platelets.Citation13,Citation14 However, none of these reports convincingly demonstrated the formation or presence of actual immune complexes in the sera of PTP patients.
The second theory proposing that patient platelets might passively absorb HPA-1a from transfused donor plasma is supported by the work of Kickler et al who showed HPA-1b/1b platelets could acquire HPA-1a following incubation with platelet-cleared plasma from stored HPA-1a donor blood. HPA-1b/1b platelets treated in this way were shown to bind anti-HPA-1a antibodies from two different sources.Citation15 However, this phenomenon has never been documented to occur in vivo. In addition, patients can develop PTP after transfusion of a single unit of red cells and can be thrombocytopenic for several weeks.Citation2 These observations bring into question how soluble transfused HPA-1a could persist in a patient’s plasma long enough to sustainably coat autologous platelets despite the presence of a potent anti-HPA-1a antibody that would be expected to promote its clearance.
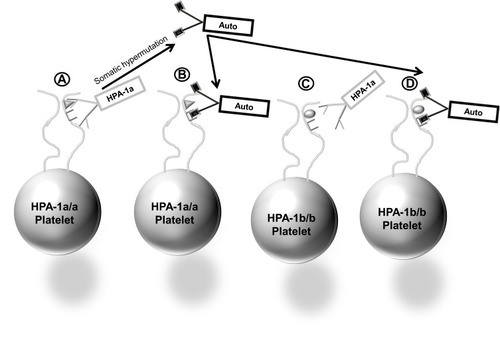
The third mechanism, proposing that platelet-specific autoantibodies produced along with one or more alloantibodies are the cause of autologous platelet destruction, is supported by several lines of evidence: 1) autologous platelets collected when the patient is thrombocytopenic contain elevated levels of platelet-associated IgG (PAIgG),Citation16,Citation17 2) clinical features and response to treatments are similar for PTP and Immune Thrombocytopenic Purpura (ITP),Citation18–Citation20 3) platelet autoantibodies are detected in the acute-phase plasma of PTP patients, but are not detectable when the patient’s platelet count recovers, whereas alloantibodies usually persist,Citation16,Citation21–Citation23 4) transfusions of HPA-1b/b platelets to PTP patients with anti-HPA-1a are cleared.Citation24 A recent report of a single case of PTP by Falk et alCitation16 in which post-recovery autologous platelets were tested against multiple serum samples obtained during the acute thrombocytopenic and post-recovery periods provides particularly convincing evidence of the autoantibody mechanism for PTP. In this study, acute-phase sera were collected at days 7, 11, 13 and 23 after surgery, and a post-recovery sample collected at day 133 post-surgery when the platelet count had normalized. Modified antigen capture enzyme-linked immunosorbent assay (MACE) testing showed that her sera had strong IgG antibodies against HPA-1a and HPA-2a expressed on platelet glycoprotein (GP) complexes GPIIb/IIIa and GPIb/IX, respectively. Her platelet genotype was HPA-1b/1b, HPA-2b/2b confirming her ability to make both alloantibodies. Flow cytometry studies of acute phase and post-recovery sera tested against autologous platelets showed auto-antibody reactivity on days 8, 11 and 13 when she was thrombocytopenic, weak auto-reactivity day 7 as her platelet count was trending down and no reactivity day 133 post recovery (). Similar findings were made by MACE showing that her acute phase serum gave positive reactions against her autologous GPIIb/IIIa.Citation16 These findings provide convincing evidence that the patient produced autoantibodies against GPIIb/IIIa, the same GP targeted by the potent HPA-1a alloantibody in her serum. The authors go on to speculate that the transition from alloantibody to autoantibody production causing PTP might be the result of somatic hypermutation of the HPA alloantibody resulting in changes to the antibody complementarity-determining region (CDR) and autoantibody formationCitation16 ().
Figure 1 Presence of platelet autoantibodies correlates with development of thrombocytopenia. An IgG antibody reactive with autologous platelets (red bars) was detected in PTP patient serum collected on Days 7, 8, 11, 13, 23 post-surgery when patient had thrombocytopenia (black line). Serum collected Day 133 post-surgery following platelet count recovery showed no autoantibodies. Strong IgG antibodies against HPA-1a/1a normal donor platelets (blue bars) were detected in sera from each time point. Values shown are flow cytometry median fluorescence intensity (MFI). Dashed line is MFI cut-off for antibody positive results.
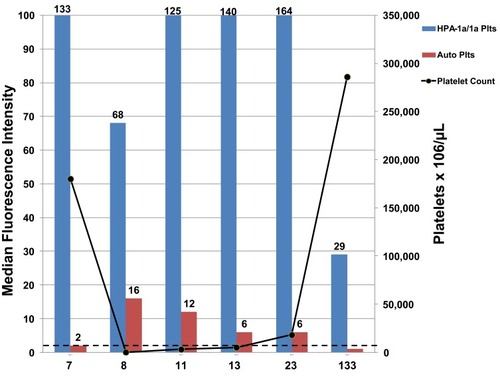
Figure 2 Cartoon depiction of autoantibody formation through somatic hypermutation of B-cell genes encoding IgG HPA-1a alloantibody (A). Autoantibody can bind platelets that are HPA-1a+ (B) or platelets that are. HPA-1a-negative (D), whereas HPA-1a antibodies can bind only HPA-1a+ platelets (A), but cannot bind and clear HPA-1a-negative platelets (C).
It is likely that platelet autoantibodies often go undetected in serologic workups of PTP patients since they are present at very low levels in the patients’ blood, probably because they are cleared in the process of destroying autologous platelets. As a result, it is often difficult to detect platelet autoantibodies in the serum of PTP patients, as is true of serum samples from many ITP patients.Citation25–Citation27
Diagnosis
Recognition of PTP can be challenging since a wide array of thrombocytopenic syndromes with symptomatic overlap can obscure the diagnosis. Severe thrombocytopenia, often <10,000 platelets/µL, 5–10 days after the inciting transfusion should raise clinical suspicion but platelet antibody testing and HPA genotyping are required to confirm. While a diagnosis of PTP can be fairly straightforward in female patients with a history of prior pregnancies and/or transfusions that received a recent blood transfusion, it can be difficult to diagnose in more complex cases, e.g. male patient with post-surgical infection having received several drugs (especially heparin) and failed multiple platelet transfusions. The differential diagnosis includes ITP, drug-induced thrombocytopenia (DITP), thrombotic thrombocytopenic purpura (TTP), heparin-induced thrombocytopenia (HIT), disseminated intravascular coagulation (DIC), sepsis and pseudothrombocytopenia. While pseudothrombocytopenia can be identified on peripheral blood smear (PBS)Citation28 and DIC typically presents with findings such as elevated PT/INR/PTT and fibrinogen consumption, the immune-mediated causes of low platelets can be difficult to delineate ().
Table 1 Findings in Immune-Mediated Thrombocytopenia
Misdiagnosis can be detrimental as each of the alternative diagnoses is a distinct entity requiring different treatment. HIT can be particularly difficult to differentiate based on clinical judgement alone, as heparin is frequently given to hospitalized patients as standard prophylaxis. Although thrombocytopenia is typically more severe in PTP, isolated thrombocytopenia can be the presenting symptom in the initial stages of HIT.Citation29 Several reported cases have highlighted the pitfalls of misdiagnosis in the setting of a recently transfused patient with a history of heparin exposure.Citation30 The goal of HIT treatment is to counteract immune complex-activated thrombosis by administering an alternative anticoagulant. This treatment regimen starkly contrasts with PTP where efforts to modulate immune function and to avoid or effectively treat bleeding are key. In this manner, a HIT misdiagnosis would delay appropriate treatment and potentially worsen PTP disease course. Concurrent laboratory testing for both HPA and heparin-PF4 antibodies is prudent.Citation2,Citation30 In all cases, but especially in complex cases or cases with significant overlap, a serologic workup for platelet antibodies provides valuable support for diagnosing PTP.
Platelet Antibody Testing
To date, 41 HPAs expressed on six different platelet glycoproteins, GPIIb, GPIIIa, GPIba, GPIbb, GPIa and CD109, have been described.Citation31 HPAs are numbered in their order of discovery with the higher frequency antigen designated “a” and the lower frequency antigen designated “b”. Twelve antigens are clustered into six biallelic groups, HPA-1, HPA-2, HPA-3, HPA-4, HPA-5 and HPA-15, and antibodies against these HPAs have been detected in the sera of PTP patients (). Both direct and indirect antibody testing can be used for detection of platelet-specific antibodies. Indirect methods test for platelet antibodies in patient plasma or serum. Direct methods are typically used to detect antibodies attached to the patient’s platelets, most often autoantibodies. Direct testing can be limited if severe thrombocytopenia is present. Glycoprotein-specific antigen capture assays are critical in identifying PLT antibodies when PTP is suspected. Enzyme-linked immunosorbent assays (ELISA) are the most widely available technique and can be used for both direct and indirect testing.
Table 2 HPA Antibodies Detected in 83 Sera from Patients with Suspected PTP
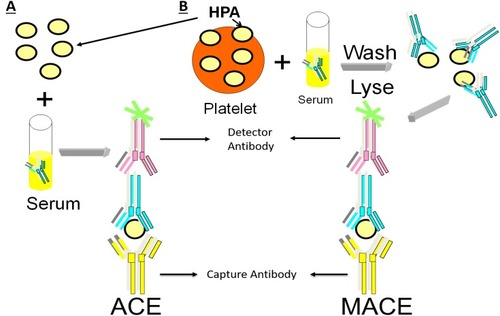
Antigen capture ELISA (ACE) and related methodologies employ a microtiter plate with platelet antigens of known specificity fixed to the plate either directly or using monoclonal antibodies. When the patient serum is added, the corresponding antibody binds its target antigen and is then detected after a second enzyme-linked anti-human Ig antibody is added ().Citation32 The widely used monoclonal antibody immobilization of platelet antigens (MAIPA) and MACE assays are variations on the ACE.Citation33–Citation35 Platelet antibody bead array (PABA) using the Luminex platform employs fluorescent magnetic beads coated with glycoprotein-specific monoclonal antibodies. Beads are incubated with a lysate of platelets sensitized by patient’s serumCitation36 or beads with captured platelet GPs are incubated directly with the patient’s serumCitation37 and HPA antibodies are then detected with fluorophore-labeled antihuman IgG by flow cytometry. Glycoprotein-specific antigen capture assays are very specific and exclude reactivity of other non-HPA antibodies which are often present in sera of multiparous women (eg. anti-HLA class I antibodies).Citation38,Citation39 If direct antibody testing is needed, washed patient platelets can be tested by MACE or MAIPA or eluted and antibody eluates tested for detection of autoantibodies.
Figure 3 ACE and MACE methods used for capture and identification of patient HPA antibodies. (A) Patient serum is incubated with HPA captured on a plate followed by enzyme-linked antibody for detection. (B) Patient serum is incubated with platelets, washed, lysed, captured and then detected using enzyme-linked antibody.
HPA Genotyping
In addition to identification of platelet antibodies, HPA typing of patient DNA is often pursued to confirm HPA antibody specificity and guide clinical care for future transfusions. Two widely used techniques include polymerase chain reaction using sequence-specific primers (PCR-SSP) and 5ʹexonuclease strategies.Citation40 More recently, HPA genotyping using a next-generation sequencing (NGS) platform has been proposed that produces large amounts of genetic data including identification of single nucleotide polymorphisms in genes that encode HPA.Citation41 This high throughput approach could lend itself to regular donor testing, creating an inventory of extensively typed blood products readily available for use by patients with antibody-mediated thrombocytopenia. However, primarily due to higher costs, NGS is not currently widely used.
Typical results in PTP cases include presence of HPA-1a antibodies in the serum and HPA-1b/1b (HPA-1a negative) genotype of the patient. Highly sensitive and specific antigen capture tests for identification of HPA-specific antibodies, like the PABACitation36 or MAIPACitation35 assays, should be performed since detection of a strong HPA antibody is the hallmark of PTP. Although HPA-1a antibodies are most frequently detected in the serum of patients suspected of PTP, sera containing platelet antibodies targeting multiple HPA are also quite common. lists the HPA antibodies detected by a large international platelet immunology reference laboratory in 83 sera from patients with suspected PTP. Interestingly, sera with HPA-1b antibodies alone (20.5%) or together with other specificities (13.2%) were the most frequently detected totaling 33.7%, and HPA-1a antibodies alone (23%) or with other specificities (9.6%) were the second most frequently detected totaling 32.6% (). A total of 22.8% of the sera contained two or more HPA-specific antibodies. This finding probably reflects the significant improvements that have been made in tests for identification of HPA antibodies. Antibodies detected against other specificities included HPA-2a, HPA-2b, HPA-3a, HPA-3b, HPA-4a, HPA-5a, HPA-5b, HPA-15b and CD36/GPIV ().
Because autoantibodies are a proposed cause of autologous platelet clearance, direct tests of patient’s platelets for platelet-associated immunoglobulins and/or platelet antigen capture tests of serum deserve consideration. Currently, such testing is rarely performed since tests for platelet autoantibodies lack sensitivity and specificity.Citation27 If testing for autoantibodies is performed, antigen capture tests (e.g. PABA, MAIPA) should be used. Evidence of platelet autoantibodies is antibody reactivity against one or more glycoproteins from every platelet tested, so-called “pan-reactivity”. Serum autoantibodies can be difficult to differentiate from the strong HPA antibodies; therefore, it is necessary to test the patient’s serum against a large panel (≥5) of Group O donor platelets expressing different combinations of HPA. Autoantibodies will be observed to give weaker reactions against glycoproteins from platelets that do not express the HPA recognized by the patient’s strongly reactive alloantibodies.
HLA antibodies are commonly detected in PTP patients’ sera, because they have been alloimmunized by previous pregnancies and/or transfusions. Although there are reports of PTP believed to be caused by HLA antibodies,Citation42 testing for HLA antibodies is not useful in the diagnosis of PTP. In order for HLA antibodies to cause autologous platelet destruction, they would have to be auto-reactive, and to date, clinically significant auto-reactive class I HLA antibodies have not been convincingly documented. However, HLA antibodies can be an important cause of platelet transfusion refractoriness.
Blood Products Implicated
PTP has been reported to be induced by whole blood, packed RBCs, platelets and plasma.Citation4,Citation9,Citation43 A report of post-transfusion thrombocytopenia after fresh frozen plasma (FFP) transfusionCitation44 attributed platelet loss to passive transfer of alloantibodies from a previously immunized donor. Passive transfer of platelet antibodies is a separate entity from PTP with different pathogenesis and disease course. In this setting, platelet nadir occurs within hours (median of 6 hrs) rather than days and recovery is often achieved within days (median of 5 days) rather than weeks.Citation44
In the literature, the incidence of PTP is variable.Citation2,Citation3 Data from the Serious Hazards of Transfusion Trial (SHOT) hemovigilance program suggest a higher frequency of PTP than previously thought. Prior to 2000, 9 to 11 cases of PTP per year were reported in the UK. That number has declined to 1 to 3 cases per year with the introduction of leukoreduction (LR).Citation45 The definitive explanation for this observation is unknown. However, Williamson et al elucidated the impact of LR on PTP by stratifying data from the SHOT study into pre and post universal LR categories.Citation9 A total of forty-five cases of PTP were reported, forty-four of which were previously pregnant females. The remaining case was found in a transfused male. Thirty-one cases were reported between1996–1999 (10.3 per year) and 14 cases (2.3 per year) were reported from 2000 to 2005 after the transition to universal LR. Studies have shown that LR of whole blood reduces PLT contamination 100-fold.Citation46 Furthermore, the authors propose that whole blood processing via the buffy coat method with subsequent LR reduces platelets to below detectable levels. A recent study by Almizraq et al demonstrated significant variability of residual platelets in red cell units based on processing methods. Notably, the average number of residual platelets was less than 1 x 103 in whole blood and apheresis-derived filtered products.Citation47 It has been theorized that the level of PLT antigens needed to induce PTP is not high enough in LR RBCs to provoke antibody formation. However, conclusive data supporting this theory are very limited. Furthermore, it is unclear what concentration of platelets is needed to induce PTP or whether different characteristics of components such as processing method, pathogen reduction or age of units pose higher risk.
It should be noted that with the re-emergence of whole blood use for trauma, increased exposure to platelet antigens should be considered. Reportedly, a unit of whole blood delivers more than double the number of platelets received during transfusion with components given at a 1:1:1 ratio; which is a widely used approach at many trauma centers.Citation48 In 2010, up to 15% of US and Canadian children’s hospitals reported using whole blood.Citation49 As of 2018, 19 major trauma centers have adopted this strategy and studies have shown similar outcomes compared to conventional components.Citation50,Citation51 As this practice gains traction, there is theoretically more platelet exposure and therefore more potential for immunization to HPA.
Treatment
Intravenous immunoglobulin (IVIG) is currently the favored treatment of choice for PTP.Citation19 Recommended doses are 400–500 mg/kg/day for 1–10 days or 1–2 gm/kg/day for 2–5 days.Citation2,Citation19,Citation23,Citation52–Citation54 Patients often have favorable response to IVIG treatment within 2 days. A 90% response rate has been reported.Citation7 Corticosteroids are often given,Citation55–Citation57 but their effectiveness is unproved.Citation19,Citation57 Whole blood or plasma exchange was the first efficacious therapy for PTP,Citation4,Citation58 and should be considered for complex cases and for cases refractory to IVIG because this treatment can be expected to quickly reduce the levels of presumptive autoantibodies. Left untreated, thrombocytopenia usually resolves within approximately 20 days.Citation57 Platelet transfusions are rarely effective at increasing platelet counts even when platelet units lacking the targeted HPA are given.Citation24,Citation59 However, platelets should be administered to patients with life-threatening bleeding.
Recurrence and Future Management
Recurrence of PTP following a subsequent transfusion is unusual.Citation6,Citation60,Citation61 Interestingly, in their original description of PTP, Shulman and colleagues repeatedly infused one of their patients with HPA-1a antibodies obtained when she was acutely thrombocytopenic. The patient did not develop thrombocytopenia or other symptoms of PTP, even after transfusions of multiple units of HPA-1a/1a-positive whole blood, platelet-rich plasma or 91 mL of her own acute plasma containing anti-HPA-1a.Citation4 Although recurrence is uncommon, previously affected patients requiring transfusion should ideally receive HPA-compatible blood products. Autologous or washed allogeneic red cells are also options. However, there is a single report of recurrence of PTP precipitated by transfusion of a deglycerolized frozen red cell product.Citation62 It has also been suggested that patients having experienced PTP should consider wearing a medic alert bracelet.Citation63
Conclusion
Since the term post-transfusion purpura was coined in 1961, over 250 cases have been reported in the literature,Citation64 yet PTP is still considered a relatively rare hazard of blood transfusion. Acute, severe thrombocytopenia that develops 5–10 days following a blood transfusion, together with primarily mucocutaneous bleeding and presence of potent HPA-specific platelet antibodies, is diagnostic of PTP. Although HPA-1a antibodies have been described most often, improvements in platelet antibody testing show that antibodies specific for HPA-1b and those targeting multiple HPA are common. Platelet autoantibody formation may play a key role in autologous platelet destruction; however, further investigation is needed. Treatment with IVIG has been standardized and has been shown to be effective. Although recurrence is rare, future precautions regarding transfusion should be taken in patients with a prior history of PTP.
Disclosure
The authors report no conflicts of interest in this work.
References
- Savage W, Hod EA. Noninfectious complications of blood transfusion In: Fung M, editor. AABB Technical Manual. 19th ed. Bethesda, MD: AABB; 2017:569–597.
- Shtalrid M, Shvidel L, Vorst E, Weinmann EE, Berrebi A, Sigler E. Post-transfusion purpura: a challenging diagnosis. Isr Med Assoc J. 2006;8(10):672–674.17125110
- Padhi P, Parihar GS, Stepp J, Kaplan R. Post-transfusion purpura: a rare and life-threatening aetiology of thrombocytopenia. BMJ Case Rep. 2013;2013:bcr2013008860.
- Shulman NR, Aster RH, Leitner A, Hiller MC. Immunoreactions involving platelets. V. Post-transfusion purpura due to a complement-fixing antibody against a genetically controlled platelet antigen. A proposed mechanism for thrombocytopenia and its relevance in “autoimmunity”. J Clin Invest. 1961;40(9):1597–1620. doi:10.1172/JCI10438316695867
- Menis M, Forshee RA, Anderson SA, et al. Posttransfusion purpura occurrence and potential risk factors among the inpatient US elderly, as recorded in large medicare databases during 2011 through 2012. Transfusion. 2015;55(2):284–295. doi:10.1111/trf.v55.225065878
- Taaning E, Svejgaard A. Post-transfusion purpura: a survey of 12 Danish cases with special reference to immunoglobulin G subclasses of the platelet antibodies. Transfus Med. 1994;4(1):1–8. doi:10.1111/j.1365-3148.1994.tb00236.x
- Padmanabhan A, Connelly-Smith L, Aqui N, et al. Guidelines on the use of therapeutic apheresis in clinical practice – evidence-based approach from the writing committee of the american society for apheresis: the eighth special issue. J Clin Apher. 2019;34(3):171–354.31180581
- Lynce F, Yin F, Alcorn K, Malkovska V. Post-transfusion purpura in an African-American man due to human platelet antigen-5b alloantibody: a case report. J Med Case Rep. 2012;6:420. doi:10.1186/1752-1947-6-42023234542
- Williamson LM, Stainsby D, Jones H, et al. The impact of universal leukodepletion of the blood supply on hemovigilance reports of posttransfusion purpura and transfusion-associated graft-versus-host disease. Transfusion. 2007;47(8):1455–1467. doi:10.1111/trf.2007.47.issue-817655590
- Rahman M, Ortega-Lopez A, Powers A. Sudden development of thrombocytopenia after reversal of anticoagulation for surgery. Lab Med. 2016;47(1):48–51. doi:10.1093/labmed/lmv00526715615
- Van Loghem JJ, Dorfmeijer H, Van Hart M, Schreuder F. Serological and genetical studies on a platelet antigen (Zw). Vox Sang. 1959;4(2):161–169. doi:10.1159/00047846513669430
- Shulman NR. Immunoreactions involving platelets. I. A steric and kinetic model for formation of a complex from a human antibody, quinidine as a haptene, and platelets; and for fixation of complement by the complex. J Exp Med. 1958;107(5):665–690. doi:10.1084/jem.107.5.66513525578
- Pegels JG, Bruynes EC, Engelfriet CP, von dem Borne AE. Post-transfusion purpura: a serological and immunochemical study. Br J Haematol. 1981;49(4):521–530. doi:10.1111/j.1365-2141.1981.tb07260.x7198482
- Lillicrap DP, Ford PM, Giles AR. Prolonged thrombocytopenia in post-transfusion purpura (PTP) associated with changes in the crossed immunoelectrophoretic pattern of von Willebrand factor (vWF), circulating immune complexes and endothelial cell cytotoxicity. Br J Haematol. 1986;62(1):37–46. doi:10.1111/j.1365-2141.1986.tb02898.x3484637
- Kickler TS, Ness PM, Herman JH, Bell WR. Studies on the pathophysiology of posttransfusion purpura. Blood. 1986;68(2):347–350. doi:10.1182/blood.V68.2.347.3473524706
- Falk G, Winans CG, Bowens K, Bougie DW, Curtis BR, Aster RH. An unexpected development after surgery-post-transfusion purpura! Am J Hematol. 2016;91(8):848–851. doi:10.1002/ajh.2441427159228
- von dem Borne AE, van der Plas-van Dalen CM. Further observation on post-transfusion purpura (PTP). Br J Haematol. 1985;61(2):374–375. doi:10.1111/j.1365-2141.1985.tb02838.x4041380
- Cunningham CC, Lind SE. Apparent response of refractory post-transfusion purpura to splenectomy. Am J Hematol. 1989;30(2):112–113. doi:10.1002/(ISSN)1096-86522913759
- McFarland J. Post transfusion purpura In: Popovsky M, editor. Transfusion Reactions. 4th ed. Bethesda, MD: AABB Press; 2012:263–287.
- Neunert CE. Management of newly diagnosed immune thrombocytopenia: can we change outcomes? Blood Adv. 2017;1(24):2295–2301. doi:10.1182/bloodadvances.201700986029296878
- Minchinton RM, Cunningham I, Cole-Sinclair M, Van der Weyden M, Vaughan S, McGrath KM. Autoreactive platelet antibody in post transfusion purpura. Aust N Z J Med. 1990;20(2):111–115. doi:10.1111/imj.1990.20.issue-22188642
- Taaning E, Tonnesen F. Pan-reactive platelet antibodies in post-transfusion purpura. Vox Sang. 1999;76(2):120–123. doi:10.1046/j.1423-0410.1999.7620120.x10085529
- Berney SI, Metcalfe P, Wathen NC, Waters AH. Post-transfusion purpura responding to high dose intravenous IgG: further observations on pathogenesis. Br J Haematol. 1985;61(4):627–632. doi:10.1111/j.1365-2141.1985.tb02876.x4084453
- Gerstner JB, Smith MJ, Davis KD, Cimo PL, Aster RH. Posttransfusion purpura: therapeutic failure of PlAl-negative platelet transfusion. Am J Hematol. 1979;6(1):71–75. doi:10.1002/(ISSN)1096-8652572138
- Brighton TA, Evans S, Castaldi PA, Chesterman CN, Chong BH. Prospective evaluation of the clinical usefulness of an antigen-specific assay (MAIPA) in idiopathic thrombocytopenic purpura and other immune thrombocytopenias. Blood. 1996;88(1):194–201. doi:10.1182/blood.V88.1.194.1948704174
- Warner MN, Moore JC, Warkentin TE, Santos AV, Kelton JG. A prospective study of protein-specific assays used to investigate idiopathic thrombocytopenic purpura. Br J Haematol. 1999;104(3):442–447. doi:10.1046/j.1365-2141.1999.01218.x10086776
- Davoren A, Bussel J, Curtis BR, Moghaddam M, Aster RH, McFarland JG. Prospective evaluation of a new platelet glycoprotein (GP)-specific assay (PakAuto) in the diagnosis of autoimmune thrombocytopenia (AITP). Am J Hematol. 2005;78(3):193–197. doi:10.1002/(ISSN)1096-865215726595
- Tan GC, Stalling M, Dennis G, Nunez M, Kahwash SB. Pseudothrombocytopenia due to platelet clumping: a case report and brief review of the literature. Case Rep Hematol. 2016;2016:3036476. doi:10.1155/2016/303647628044112
- Linkins LA, Dans AL, Moores LK, et al. Treatment and prevention of heparin-induced thrombocytopenia: antithrombotic therapy and prevention of thrombosis, 9th ed: American college of chest physicians evidence-based clinical practice guidelines. Chest. 2012;141(2Suppl):e495S–e530S. doi:10.1378/chest.11-230322315270
- Lubenow N, Eichler P, Albrecht D, et al. Very low platelet counts in post-transfusion purpura falsely diagnosed as heparin-induced thrombocytopenia. Report of four cases and review of literature. Thromb Res. 2000;100(3):115–125. doi:10.1016/S0049-3848(00)00311-X11108897
- Human Platelet Antigen (HPA) database. Versiti. Available from: https://www.versiti.org/medical-professionals/precision-medicine-expertise/platelet-antigen-database#hpa-database.Accessed 1029, 2019.
- Heikal NM, Smock KJ. Laboratory testing for platelet antibodies. Am J Hematol. 2013;88(9):818–821. doi:10.1002/ajh.v88.923757218
- Kim H, Oh EJ, Kim J, Park YJ, Han K. Detection of platelet specific antibodies by modified antigen capture ELISA test. Korean J Lab Med. 2006;26(3):192–197. doi:10.3343/kjlm.2006.26.3.19218156724
- Lochowicz AJ, Curtis BR. Clinical applications of platelet antibody and antigen testing. Lab Med. 2011;42(11):687–692. doi:10.1309/LMM242GKGHCKJRWI
- Kiefel V, Santoso S, Weisheit M, Mueller-Eckhardt C. Monoclonal antibody–specific immobilization of platelet antigens (MAIPA): a new tool for the identification of platelet-reactive antibodies. Blood. 1987;70(6):1722–1726. doi:10.1182/blood.V70.6.1722.17222445398
- Metzner K, Bauer J, Ponzi H, Ujcich A, Curtis BR. Detection and identification of platelet antibodies using a sensitive multiplex assay system-platelet antibody bead array. Transfusion. 2017;57(7):1724–1733. doi:10.1111/trf.2017.57.issue-728439930
- Porcelijn L, Huiskes E, Comijs-van Osselen I, et al. A new bead-based human platelet antigen antibodies detection assay versus the monoclonal antibody immobilization of platelet antigens assay. Transfusion. 2014;54(6):1486–1492. doi:10.1111/trf.1250924299453
- Kakaiya RM, Triulzi DJ, Wright DJ, et al. Prevalence of HLA antibodies in remotely transfused or alloexposed volunteer blood donors. Transfusion. 2010;50(6):1328–1334. doi:10.1111/trf.2010.50.issue-620070615
- Triulzi DJ, Kleinman S, Kakaiya RM, et al. The effect of previous pregnancy and transfusion on HLA alloimmunization in blood donors: implications for a transfusion-related acute lung injury risk reduction strategy. Transfusion. 2009;49(9):1825–1835. doi:10.1111/j.1537-2995.2009.02206.x19453983
- Curtis BR, McFarland JG. Human platelet antigens – 2013. Vox Sang. 2014;106(2):93–102. doi:10.1111/vox.1208524102564
- Davey S, Navarrete C, Brown C. Simultaneous human platelet antigen genotyping and detection of novel single nucleotide polymorphisms by targeted next-generation sequencing. Transfusion. 2017;57(6):1497–1504. doi:10.1111/trf.2017.57.issue-628370162
- Vaughan-Neil EF, Ardeman S, Bevan G, Blakeman AC, Jenkins WJ. Post-transfusion purpura associated with unusual platelet antibody (anti-Pl-B1). Br Med J. 1975;1(5955):436–437. doi:10.1136/bmj.1.5955.4361115962
- McCrae KR, Herman JH. Posttransfusion purpura: two unusual cases and a literature review. Am J Hematol. 1996;52(3):205–211. doi:10.1002/(ISSN)1096-86528756089
- Pavenski K, Webert KE, Goldman M. Consequences of transfusion of platelet antibody: a case report and literature review. Transfusion. 2008;48(9):1981–1989. doi:10.1111/trf.2008.48.issue-918564398
- Bolton-Maggs PH, Cohen H. Serious hazards of transfusion (SHOT) haemovigilance and progress is improving transfusion safety. Br J Haematol. 2013;163(3):303–314. doi:10.1111/bjh.1254724032719
- Prowse CV, Hornsey VS, Drummond O, et al. Preliminary assessment of whole-blood, red-cell and platelet-leucodepleting filters for possible induction of prion release by leucocyte fragmentation during room temperature processing. Br J Haematol. 1999;106(1):240–247. doi:10.1046/j.1365-2141.1999.01530.x10444194
- Almizraq RJ, Norris PJ, Inglis H, et al. Blood manufacturing methods affect red blood cell product characteristics and immunomodulatory activity. Blood Adv. 2018;2(18):2296–2306. doi:10.1182/bloodadvances.201802193130217795
- Pivalizza EG, Stephens CT, Sridhar S, et al. Whole blood for resuscitation in adult civilian Trauma in 2017: a narrative review. Anesth Analg. 2018;127(1):157–162. doi:10.1213/ANE.000000000000342729771715
- Spinella PC, Dressler A, Tucci M, et al. Survey of transfusion policies at US and Canadian children’s hospitals in 2008 and 2009. Transfusion. 2010;50(11):2328–2335. doi:10.1111/trf.2010.50.issue-1120529008
- Holcomb JB, Jenkins DH. Get ready: whole blood is back and it’s good for patients. Transfusion. 2018;58(8):1821–1823. doi:10.1111/trf.1481830144081
- Seheult JN, Anto V, Alarcon LH, Sperry JL, Triulzi DJ, Yazer MH. Clinical outcomes among low-titer group O whole blood recipients compared to recipients of conventional components in civilian trauma resuscitation. Transfusion. 2018;58(8):1838–1845. doi:10.1111/trf.1477930160310
- Mueller-Eckhardt C, Kuenzlen E, Thilo-Korner D, Pralle H. High-dose intravenous immunoglobulin for post-transfusion purpura. N Engl J Med. 1983;308(5):287.
- Becker T, Panzer S, Maas D, et al. High-dose intravenous immunoglobulin for post-transfusion purpura. Br J Haematol. 1985;61(1):149–155. doi:10.1111/j.1365-2141.1985.tb04071.x4052323
- Hamblin TJ, Naorose Abidi SM, Nee PA, Copplestone A, Mufti GJ, Oscier DG. Successful treatment of post-transfusion purpura with high dose immunoglobulins after lack of response to plasma exchange. Vox Sang. 1985;49(2):164–167. doi:10.1159/0004663654036084
- Slichter SJ. Post-transfusion purpura: response to steroids and association with red blood cell and lymphocytotoxic antibodies. Br J Haematol. 1982;50(4):599–605. doi:10.1111/j.1365-2141.1982.tb01960.x7199931
- Christie DJ, Pulkrabek S, Putnam JL, Slatkoff ML, Pischel KD. Posttransfusion purpura due to an alloantibody reactive with glycoprotein Ia/IIa (anti-HPA-5b). Blood. 1991;77(12):2785–2789. doi:10.1182/blood.V77.12.2785.27852043772
- Vogelsang G, Kickler TS, Bell WR. Post-transfusion purpura: a report of five patients and a review of the pathogenesis and management. Am J Hematol. 1986;21(3):259–267. doi:10.1002/(ISSN)1096-86523946408
- Cimo PL, Aster RH. Post-transfusion purpura: successful treatment by exchange transfusion. N Engl J Med. 1972;287(6):290–292. doi:10.1056/NEJM1972081028706085038955
- Brecher ME, Moore SB, Letendre L. Posttransfusion purpura: the therapeutic value of PlA1-negative platelets. Transfusion. 1990;30(5):433–435. doi:10.1046/j.1537-2995.1990.30590296377.x2360235
- Budd JL, Wiegers SE, O’Hara JM. Relapsing post-transfusion purpura. A preventable disease. Am J Med. 1985;78(2):361–362. doi:10.1016/0002-9343(85)90451-64038575
- Puig N, Sayas MJ, Montoro JA, Villalba JV, Pla A. Post-transfusion purpura as the main manifestation of a trilineal transfusion reaction, responsive to steroids: flow-cytometric investigation of granulocyte and platelet antibodies. Ann Hematol. 1991;62(6):232–234. doi:10.1007/BF017298391854887
- Godeau B, Fromont P, Bettaieb A, Duedari N, Bierling P. Post-transfusion purpura. An unknown cause of acute immune thrombocytopenia. 4 new cases]. Presse Med. 1990;19(43):1974–1977.2149598
- Vincent EC, Willett T. Post-transfusion purpura. J Am Board Fam Pract. 1991;4(3):175–177.1810275
- Immunologic transfusion reactions. Eds. Jennifer S Tirnauer; 2019 Available from: https://www.uptodate.com/contents/immunologic-transfusion-reactions?search=post%20transfusion%20purpura§ionRank=1&usage_type=default&anchor=H18&source=machineLearning&selectedTitle=1~12&display_rank=1#H18. Accessed 715, 2019.
- Page EE, Kremer Hovinga JA, Terrell DR, Vesely SK, George JN. Thrombotic thrombocytopenic purpura: diagnostic criteria, clinical features, and long-term outcomes from 1995 through 2015. Blood Adv. 2017;1(10):590–600. doi:10.1182/bloodadvances.201700512429296701
- Warkentin TE. Low-molecular weight heparins in prophylaxis and therapy of thromboembolic diseases In: Bounameaux H, editor. Low-Molecular Weight Heparins in Prophylaxis and Therapy of Thromboembolic Diseases. New York: Marcel Dekker; 1994:75.
- Warkentin TE. Clinical presentation of heparin-induced thrombocytopenia. Semin Hematol. 1998;35(4Suppl 5):9–16.
- Ortel TL. Heparin-induced thrombocytopenia: when a low platelet count is a mandate for anticoagulation. Hematology Am Soc Hematol Educ Program. 2009;2009:225–232. doi:10.1182/asheducation-2009.1.225
- Aster RH, Curtis BR, McFarland JG, Bougie DW. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis, and management. J Thromb Haemost. 2009;7(6):911–918. doi:10.1111/jth.2009.7.issue-619344362
- Scully M, Yarranton H, Liesner R, et al. Regional UK TTP registry: correlation with laboratory ADAMTS 13 analysis and clinical features. Br J Haematol. 2008;142(5):819–826. doi:10.1111/bjh.2008.142.issue-518637802
- Jamme M, Rondeau E. The PLASMIC score for thrombotic thrombocytopenic purpura. Lancet Haematol. 2017;4(4):e148–e149. doi:10.1016/S2352-3026(17)30024-828259521
- Bendapudi PK, Hurwitz S, Fry A, et al. Derivation and external validation of the PLASMIC score for rapid assessment of adults with thrombotic microangiopathies: a cohort study. Lancet Haematol. 2017;4(4):. doi:10.1016/S2352-3026(17)30026-1
- Lo GK, Juhl D, Warkentin TE, Sigouin CS, Eichler P, Greinacher A. Evaluation of pretest clinical score (4 T’s) for the diagnosis of heparin-induced thrombocytopenia in two clinical settings. J Thromb Haemost. 2006;4(4):759–765. doi:10.1111/jth.2006.4.issue-416634744
- Cuker A, Gimotty PA, Crowther MA, Warkentin TE. Predictive value of the 4Ts scoring system for heparin-induced thrombocytopenia: a systematic review and meta-analysis. Blood. 2012;120(20):4160–4167. doi:10.1182/blood-2012-07-44305122990018
- Neunert C, Lim W, Crowther M, et al. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood. 2011;117(16):4190–4207. doi:10.1182/blood-2010-08-30298421325604
- Al-Samkari H, Kuter DJ. Optimal use of thrombopoietin receptor agonists in immune thrombocytopenia. Ther Adv Hematol. 2019;10:2040620719841735. doi:10.1177/204062071984173531007886
- Scully M, Cataland SR, Peyvandi F, et al. Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura. N Engl J Med. 2019;380(4):335–346. doi:10.1056/NEJMoa180631130625070
- Cuker A, Arepally GM, Chong BH, et al. American Society of Hematology 2018 guidelines for management of venous thromboembolism: heparin-induced thrombocytopenia. Blood Adv. 2018;2(22):3360–3392. doi:10.1182/bloodadvances.201802448930482768