Abstract
Congenital Factor XIII (FXIII) deficiency is a rare, inherited, autosomal recessive coagulation disorder. Most mutations of this condition are found in the A-subunit with almost half these being missense mutations. Globally, approximately one in three million people suffer from this deficiency. Factor XIII deficiency is associated with severe life threatening bleeding, intracranial hemorrhage, impaired wound healing, and recurrent pregnancy losses. FXIII is known to have a potential role in mediating inflammatory processes, insulin resistance, bone metabolism, neoplasia, and angiogenesis. The algorithm provided for FXIII diagnosis and classification will enable prompt identification and early intervention for controlling potential life threatening complications. Prophylactic replacement therapy using blood products containing FXIII such as fresh frozen plasma, cryoprecipitate, or using FXIII concentrate remains the mainstay for the management of FXIII deficiency. In most parts of the world, cryoprecipitate and plasma transfusions are the only treatments available. Management developments have revealed the effectiveness and safety of recombinant FXIII concentrate for prophylaxis and treatment. The aim of this review is to provide an overview of advancements made in the management of FXIII deficiency from the time it was first detected, highlighting novel developments made in recent years. Greater research is warranted in identifying novel approaches to manage FXIII deficiency in light of its underlying pathophysiology.
Introduction
Approximately one in every three to five million people suffer from congenital Factor XIII (FXIII) deficiency globally. This coagulation blood disorder is inherited in an autosomal recessive manner.Citation1 The incidence of the disease in the United Kingdom is known to be one in every two to five million people.Citation2,Citation3 The disease is so rare that, to date, a little over 300 cases have been reported worldwide.Citation4 The greatest number of cases have been reported in Japan.Citation5,Citation6 This rare bleeding disorder affects people of all races and genders with a higher prevalence in families with FXIII deficient patients and consanguinity. In the case of nonconsanguineous families, a higher incidence of compound heterozygosity is observed.Citation7–Citation10 High rates of consanguineous marriages are seen in South Asia.Citation11 A cross-section study conducted at a blood bank in Pakistan identified nine cases of Factor XIII through routine screening over a 7 year period.Citation12 A recent case-series report at a tertiary care hospital in the region identified ten cases of FXIII deficiency over a 10 year period with 80% of cases having a history of consanguineous marriages.Citation5
The clinical importance of FXIII came to light in the year 1960 when a boy in Switzerland was reported to have suffered from a severe bleeding diathesis 16 years after its discovery. The only bleeding abnormality described by Duckert et al was the solubility of his clots in 5Molar urea (concentration).Citation13
Most congenital FXIII deficiencies are a result of FXIII-A subunit deficiency.Citation14 The A subunit in the inherited deficiency is absent from plasma, platelets, and monocytes. Roughly 50% of molecular defects that cause a deficiency in the FXIII-A subunit are missense mutations.Citation15 On the other hand, congenital deficiency in the FXIII-B subunit is a relatively rarer cause of FXIII deficiency.Citation15 Plasma levels of the B subunit are usually reduced, and very rarely both A and B subunits are absent.Citation7 Bleeding symptoms in patients with FXIII-B deficiencies are relatively mild compared to FXIII-A deficiencies.Citation16 Severe clinical symptoms have been reported in fewer than ten cases.Citation16
Molecular structure
FXIII in the plasma (pFXIII) is a precursor of transglutaminase and is a tetrameric complex (FXIII-A2B2) of two active A subunits (FXIIIA; molecular weight 83 kDa) and two carrier B subunits (FXIII-B; molecular weight 80 kDa). The gene coding for FXIII-A is located on chromosome 6p24-25, whereas the FXIII-B gene is located on chromosome 1 at 1q31-32.1.Citation17 FXIII-A is synthesized in cells of bone marrow origin, whereas FXIII-B is produced by hepatocytes and the two types of subunits form a complex in the plasma.Citation15 The cellular form of FXIII (cFXIII), a homodimer of FXIII-A (FXIII-A2), is found in the cytoplasm of platelets, monocytes, histiocytes, and tissue macrophages.Citation14 FXIII-B is the protective unit of the tetramer that protects subunit A from proteolysis and under normal physiological conditions is found in excess in the serum as free FXIII-B.Citation18
F XIII is the last factor in the coagulation cascade. It is a protransglutaminase, which is activated by thrombin to transglutaminase in presence of calcium ions.Citation19 Fibrin, along with optimal serum calcium levels, also functions as an important cofactor in the activation process and accelerates the activation rate manifold.Citation1,Citation20,Citation21 Activated FXIII converts loose fibrin polymers into an organized structure by cross linking the peptide-bound glutamyl and lysine residues of fibrinogen chains through an isopeptide bond, thereby releasing ammonia. This stable fibrin clot that is formed as a result adheres firmly to the underlying wound and is not easily degraded by sheer stress and the fibrinolytic system. Plasma concentrations of FXIII and age of the clot are the two most important determinants of a clot’s resistance to degradation by the fibrinolytic system. The average plasma concentration of the A2B2 heterotetramer is approximately 22 μg/mL and its half-life is 9–14 days.Citation22
A study conducted using two separate lines of FXIII-A knockout mice with prolonged bleeding times indicated impaired clot retraction in FXIII-A deficient mice in the presence of normal platelet aggregation induced by collagen and adenosine diphosphate.Citation23
In recent years, developments in DNA technology have facilitated genetic studies in families with inherited deficiencies as well as in normal individuals. The nucleotide substitutions leading to nonsense mutations, missense mutations, and splice defects do not appear to be clustered in specific regions on the gene, but are spread over the entire FXIII-A gene along with its messenger RNA. Studies focusing on the structural and functional consequences of these mutations and normal polymorphisms in the gene have translated into a better understanding of FXIII function.Citation7 These mutations predict the severity of the disease in patients deficient in FXIII.Citation24–Citation26
Classifying FXIII deficiency
Formerly, FXIII deficiency was categorized as either type 1 or type 2. Type 1 deficiency was described as a combined deficiency of FXIII-A and FXIII-B, whereas type II was defined as FXIII-A deficiency only. However, this classification is outdated as it was later learnt that patients assumed to have type I combined deficiency are in fact defective in the FXIII-B geneCitation27 and that the lower FXIII-A level is due to increased clearance from the circulation in the absence of protective FXIII-B.Citation14 Nevertheless, for the purpose of classification (), FXIII-A2B2 antigen in the plasma is first determined, and if decreased, further measurement of the individual subunits is recommended in the plasma and FXIII-A in platelet lysate.Citation14
Figure 1 Algorithm for diagnosis and classification of Factor XIII deficiency.
Reprinted from Song JW, Choi JR, Song KS, Rhee JH. Plasma factor XIII activity in patients with disseminated intravascular coagulation. Yonsei Med J. 2006;47(2):196–200.
Abbreviation: FXiii, factor Xiii.
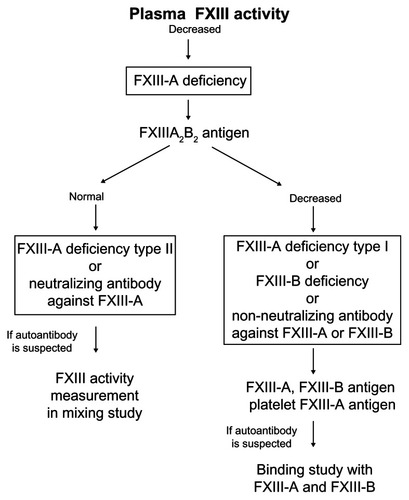
Inherited FXIII deficiency is now classified as FXIII-A and FXIII-B. FXIII-A can further be sub-classified as type 1 or type 2 defects where type 1 represents a quantitative defect, whereas type 2 represents a qualitative defect.Citation14 Although recognized as a hereditary disorder,Citation28 with research advancements, it is now known that FXIII-A deficiency is due to mutations in gene coding the catalytic A subunit (FXIII-A) located on Chromosome 6. A little over 104 mutations have been associated with this deficiency and the majority are a result of missense and nonsense mutations. B subunits are typically normal in such patients.Citation29 FXIII-B is associated with mutations in the gene encoding the B subunit located on Chromosome 1. Around 16 mutations that lead to FXIII-B deficiency have been identified, which are less common than FXIII-A (type 1 and type 2) deficiency.Citation29 However, a case of combined FXIII-A and FXIII-B deficiency has been reported as a result of an insertion mutation in the second sushi domain of the B subunit.Citation29
A number of cases of FXIII deficiency are acquired deficiencies. These occur as a result of disorders or diseases associated with its overconsumption and biosynthesis.Citation30 The deficiency is either a drug-induced or an autoimmune disorder and is most common among geriatric patients.Citation31 Acquired FXIII deficiency has also been linked to a variety of diseases including rheumatoid arthritis and systemic lupus erythematosus.Citation32–Citation34 Significant reductions in FXIII have also been reported in medical conditions like pulmonary embolism, stroke, leukemia, Crohn’s disease, ulcerative colitis, Henoch–Schönlein purpura, liver cirrhosis, sepsis, renal dysfunction, and disseminated intravascular coagulation.Citation35 Decreased hepatic synthesis reduces the half-life of the A-subunit in the absence of the B-subunit.Citation36
Clinical manifestations
Congenital FXIII deficiency can manifest as a severe bleeding conditions as early as a few days after birth in the form of umbilical stump bleeding.Citation37 The bleeding in FXIII deficient individuals is more severe as compared to other coagulative disorders. Intracutaneous bleeding (57%), umbilical stump bleeds (56%), intramuscular bleeding (49%), and intracranial hemorrhage (34%) are the most common clinical manifestations in these individuals as determined by International Registry of Factor XIII deficiency.Citation1,Citation37–Citation39 The rates of umbilical bleeding and intracranial hemorrhage are much higher compared to Hemophilia and Type III von Willebrand factor deficiency.Citation40
Most patients with congenital FXIII deficiency suffer from life-long crippling bleeding diathesis with a very high risk of early mortality. Nearly 80% of deaths are attributed to intracranial hemorrhage.Citation7 Clinical manifestations vary in patients and are unpredictable whereby long periods of mild symptoms may follow severe bleeding complications.Citation22
Another early manifestation of FXIII deficiency is post-operative bleeding, which usually occurs as a result of disruption of an unstable clot.Citation41 Circumcision in deficient individuals 24 hours after birth has resulted in severe bleeding.Citation7 Other characteristic symptoms include ecchymosis, intramuscular and subcutaneous hematomas, oral cavity, mouth and gingival bleeding, and prolonged bleeding following trauma.Citation22 Hemarthrosis occurs in a number of patients,Citation42 with joint-associated bleeding usually occurring periarticular rather than into the joint cavity.Citation7,Citation43 A rare case of subdural and epidural hematoma has also been reported in the literature deeming it necessary to screen a patient with a bleed with no identifiable cause.Citation44 Studies have shown that many patients recover if managed with adequate replacement therapy alone.Citation45 Surgery has been reserved for worsening neurological conditions where prompt intervention has had life-saving outcomes, especially for subdural bleeds.Citation46
Besides its role in hemostatic function, FXIII deficiency is also associated with poor wound healing and angiogenesis.Citation14,Citation41 Evidence from studies suggests FXIII-A2 facilitates Fcc and complement receptor mediated phagocytosis. Deficient patients have been shown to have impaired function in these activities.Citation47 Murine wound healing models also support the important role of intracellular FXIII-A2 in leukocyte and tissue remodeling and repair. Studies on myocardial repair following infarction induced in FXIII knockout mice demonstrated a reduction in leukocyte recruitment, phagocytosis, and protease activity in injured myocardial tissue.Citation48
Factor XIII is known to play a role in maintaining pregnancy.Citation49 The rate of miscarriage in FXIII deficient females can be as high as 80%.Citation41 The production of FXIII-A2 in the placenta, confirms its role in maintaining the integrity of placental attachment in the uterus.Citation50 A study assessing FXIII plasma activity in pregnancy has shown that low maternal plasma activity of FXIII leads to a low concentration of subunit A at the placental bed resulting in the formation of an insufficient and inadequate cytotrophoblastic shell and increased risk of miscarriage.Citation51 Recurrent fetal losses with a normal miscarriage workup and a family history negative for bleeding disorders needs to be evaluated for FXIII deficiency.Citation52
Molecular research focusing on the implications of FXIII deficiency in adults identified a lower prevalence of myocardial infarction in subjects without a factor XIII Val 34 Leu mutation.Citation1 A relatively higher incidence of primary intracerebral hemorrhage was seen in patients with factor XIII Val 34 Leu mutation.Citation43 However, the precise role of fibrin in causing cerebrovascular disease is unknown. Nevertheless, the formation of cross-linked fibrin from fibrin monomer is vital for the development and maintenance of a stable clot, and abnormalities of fibrin structure and architecture are associated with premature myocardial infarction in males.Citation53
New developments reveal the role of FXIII for functions other than coagulation and cardiovascular disease, maintaining pregnancy, and wound healing. FXIII is known to play a role in osteoblast matrix secretion and deposition where FXIII-A and its cross-linking activity are localized with plasma membrane-associated tubulin. The cross-linking activity is aimed at stabilizing the interaction of microtubules with the plasma membrane, demonstrating that transglutaminase activity can affect protein secretion and matrix deposition in osteoblasts.Citation54,Citation55 FXIII has been shown to have a direct role on vascular endothelial cells in promoting angiogenesis in vitro and in vivo on animal models.Citation56 Insulin resistance and diabetes mellitus is associated with prothrombotic states, atherosclerosis, dyslipidemia, and obesity. The exact mechanism of the role of FXIII in this context is unknown; however, its influence on the mentioned risk factors can lead FXIII deficient individuals to be at a higher risk of insulin resistance. Moreover, the proinflammatory effect of FXIII can exacerbate cardiovascular risks and insulin resistance.Citation57,Citation58 Induction of the coagulation cascade leads to an immune response involving the mobilization of immune-mediated cells and killing bacteria in the clot. This entrapment of bacteria is mediated by FXIII-A which cross-links bacteria to fibrin.Citation59,Citation60 Expression of FXIII may potentially be considered as a leukemia-associated immunophenotype. FXIII assays may be used for diagnosis or for monitoring disease progress.Citation61,Citation62
Diagnosis
It is important to screen patients born with a bleeding diathesis, or with a family history of bleeding disorders in order to start prophylactic therapy to lower the risk of spontaneous or acquired intracranial bleeding and its complications.Citation63 Diagnosis might be delayed in situations where a family history of bleeding disorders is not revealed.Citation64 Lack of awareness of this condition in emergency units has also been associated with delayed diagnosis.Citation65
Standard hemostasis assays, including prothrombin time, activated partial thromboplastin time, fibrinogen level, platelet count, and platelet function testing, are normal in individuals with FXIII deficiency as FXIII acts when fibrin has already been formed. These assays have proven to be effective for diagnoses of bleeding disorders, such as congenital or acquired hemophilia, where there is a deficiency in factor VIII or factor IX.Citation66 However, none of these tests are capable of detecting a deficiency in FXIII.Citation29
Since the discovery of the first case of FXIII in 1960 through standard clot solubility testing, labs have used this test for the diagnosis of FXIII deficiency. The test involves using a small volume of the patient’s plasma which is incubated at room temperature with a buffer and a solution of calcium ± thrombin. Incubation is continued for approximately 1-hour to allow for clot stabilization. The clot is then suspended in a freshly prepared solution of 1% monochloroacetic acid or in a solution of 5Molar urea. Under normal conditions, rapid dissolution of the clot should occur anywhere between a few minutes to 1-hour. The clot solubility test is a qualitative test and is only positive if FXIII activity is zero or close to zero. Adding a small amount of normal plasma to the system elevates FXIII activity to 1% ± 3% of normal, rendering the clot insoluble.Citation7
However, this test is poorly standardized and detects only very severe deficiencies in FXIII activity.Citation7,Citation29 FXIII < 1 UdL−1 has been associated with a severe bleeding risk, whereas levels of between 1 and 4 U dL−1 indicate moderate disease. Even with an accurate measure of FXIII levels, the phenotype does not always correlate with levels obtained, as those with levels > 5 U dL−1 have also suffered bleeding complications.Citation7 However, although this test is no longer found to be a reliable screening tool, it is still used by most routine laboratories because of its simplicity and the lack of availability of quantitative screening tools.Citation15
If the diagnosis of FXIII deficiency is suggested by the solubility test, other quantitative tests can be undertaken.Citation2,Citation67 Mild to moderate deficiencies in FXIII are better diagnosed through the use of a quantitative assay, such as amine incorporationCitation68–Citation71 and ammonia release assays,Citation72,Citation73 which measure the transglutaminase activity of FXIII. The screening test, which establishes the diagnosis of FXIII deficiency, should be a FXIII activity assay. FXIII activity assays are based on: (i) measuring the ammonia released during the transglutaminase reaction by the nicotinamide adenine dinucleotide phosphate (NAD[P])H-dependent glutamate dehydrogenase reaction spectrophotometrically at 340 nm, and (ii) measuring the amount of a small molecular weight labeled amine substrate covalently linked to a protein. In the latter case, the free and bound radiolabeled, fluorescent, or biotinylated amine should be separated and the protein-linked fraction is quantitatively measured.Citation14 Amine incorporation methods are known to have a high sensitivity, are automated, resulting in quick kinetic testing.Citation14 However, they are more time consuming than the ammonia release assays, difficult to standardize, and the separation step makes it difficult to design a true kinetic assay.Citation14
On the other hand, disadvantages of the ammonia test include its relatively low sensitivity.Citation14 The assay may give overestimated results in low FXIII concentrations as high ammonia levels in the test plasma compromise accuracy. Mixing studies with normal plasma can avoid this complication. Levels below 15% should be closely evaluated as they may contain either traces of FXIII or no FXIII at all.Citation39,Citation74
Enzyme-linked immunosorbent assay (ELISA) has been recommended to account for the aforementioned drawbacks of traditional testing. ELISA has been used to determine FXIII or antigen levels to diagnose or monitor therapy for FXIII deficiency. The test directly measures FXIII A-subunit or FXIII B-subunit proteins using specific antibodies. ELISA is a sensitive technique, thus making it reliable for low range FXIII levels.Citation39 shows the expected levels in different subtypes.Citation15
Table 1 Laboratory Diagnosis/Classification of factor XIII deficiency
The diagnostic tools used for the classification of FXIII deficiencies include FXIII-A2B2, FXIII-A, and FXIII-B antigen determinations from the plasma, FXIII activity, and FXIII-A antigen measurement from the platelet lysate produced using a nonionic detergent. Study of fibrin cross-linking by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS PAGE) analysis of washed plasma clot is a useful addition in a series of tests to confirm diagnoses.Citation14
It has become highly desirable to also study the type of mutations causing the defect to fully characterize the patient and contribute to our understanding of disease mechanisms.Citation14 PCR has been shown to play a role in the diagnosis of FXIII deficiency. With over 100 mutations having been identified in the gene coding for FXIII and the type of mutation affecting the presentation and disease course, genetic testing has a significant role in the diagnosis of FXIII deficiency.Citation10,Citation15,Citation71 Once the mutation has been identified it can aid in family counseling, prenatal screening, and further characterization of specific bleeding risk.Citation19
Treatment
FXIII deficiency is one of the few coagulation disorders where great emphasis is placed on primary prophylaxis because of severe life threatening complications. Prophylactic treatment (10/20 U/kg FXIII every 4–6 weeks) is recommended for all patients diagnosed with severe FXIII deficiency to prevent life-threatening bleeds and intracranial hemorrhage. It is strongly recommended for patients with serum levels of FXIII below 1 U/dL at diagnosis to receive prophylaxis as this group is at greatest vulnerability to severe bleeding complications.Citation28 Patients with a serum FXIII level between 1–4 U/dL are also prone to moderate to severe spontaneous bleeding episodes and prophylactic replacement therapy should also be considered in this subset of deficient individuals.Citation75 Serum FXIII levels > 5 U/dL are desirable but do not rule out the possibility of spontaneous bleeds that may occur in a small percentage of patients.Citation38
Despite the rarity of the disease, studies have focused on long term effects of prophylactic treatment. Two prospective studies demonstrated that prophylactic treatment successfully kept a check on bleeding episodes, without any significant toxicity or transmission of infection.Citation76
Evidence from the literature suggests that FXIII levels of 3%–10% of the normal population mean (0.03–0.1 IU/mL) are sufficient to prevent spontaneous bleeds.Citation77,Citation78 A recent data analysis concluded that a plasma concentration of 10% is needed to significantly prevent episodes of any spontaneous hemorrhages. However, there may still remain a chance of cutaneous bleeds in 10% of patients.
Even though plasma derived sources of FXIII, namely, whole blood, fresh frozen plasma, and cryoprecipitate have been widely used for decades due to the ease of availability, they carry a high risk for allergic reaction and infections with blood-borne pathogens, including hepatitis and human immunodeficiency virus (HIV). Highly purified and heat treated FXIII concentrate from plasma is the treatment of choice for long term prophylaxis as it contains adequate and reliable concentrations of FXIII in optimal volume with fewer contaminations and is virally inactivated.Citation1 FXIII concentrate has the longest plasma half-life (11–14 days) of all clotting factors. Primary prophylaxis is practical and feasible in cases of FXIII deficiency as the concentrate has a long half-life and only a low plasma concentration of 10% is needed to significantly reduce bleeding episodes.
Since placenta derived FXIII was withdrawn from the market in 1994, plasma derived pasteurized concentrate of FXIII marketed under the name of Fibrogammin P has been widely used for prophylaxis. It is licensed for use in several countries in Asia, South America, Europe while phase II/III trials are still ongoing in United States to assess its efficacy and safety profile. Fibrogammin P is derived from HIV negative pooled human plasma screened for common viruses (hepatitis B surface antigen, anti-hepatitis C virus, anti-HIV-1, and anti-HIV-2). It is available in pharmacies as a 250 IU pack for IV administration for £106.58 (£0.42/Unit) and is equally efficacious in all forms of congenital FXIII deficiency (A and B). An ongoing investigational new drug study in the United States, evaluating the prophylactic efficacy and long-term safety of Fibrogammin P has 61 enrolled subjects (Male 44, Female 17; Mean age 12.7); representing approximately two-thirds of patients with FXIII deficiency nationwide.Citation79 Response to therapy with a 9-year follow up has been good to excellent, without any evidence of FXIII inhibitor development or seroconversion. No major intracranial or life-threatening bleeds have been reported in these patients. The half-life of the concentrate in serum is comparable to that of patients who receive cryoprecipitate or fresh frozen plasma.Citation79
For patients with congenital factor XIII deficiency that undergo any major surgery, it is required that a higher dose of 20–30 U/kg/day be administered instead of the prophylactic dose (10–20 U/kg every 4–6 weeks), to maintain a plasma concentration of higher than 5%.Citation80 Replacement therapy should ideally be administered immediately prior to surgery and should be continued until complete recovery is made. A dose of 10–20 U/kg/day for 2–3 days should be sufficient for minor surgeries.Citation80
In acute and severe bleeding conditions, a FXIII concentrate can be administered at a dose of 20 U/kg. Further treatment depends on serum FXIII levels achieved after administering the initial dose. FXIII infusion should continue until bleeding stops. In case a patient develops intracranial hemorrhage, FXIII levels need to be monitored closely and maintained at a normal range for a minimum of 2 weeks before reverting to therapy at the prophylactic dosage. Such aggressive management will require replacement therapy on alternate days.Citation80 If, for any reason, FXIII concentrate or plasma is not available in hemorrhagic emergencies, platelet transfusion can be used as an alternative means of providing hemodynamic stability as FXIII is contained in platelets.
Neonates with severe FXIII deficiency (<3 U/dL) are at a high risk of life threatening umbilical bleeding, cephalohematoma, and intracranial bleeding. Aggressive management is recommended with 10 U/kg prophylactic therapy given at 4 weekly intervals.Citation80 Subsequent dosages and frequency of doses depends upon pretreatment (<3 U/dL) and 1-hour post treatment (>60 U/dL) plasma FXIII levels.Citation80 Assuming young children are active and more prone to injury, 4 weekly FXIII replacements have been recommended to achieve a constant serum concentration of at least 10% of mean serum FXIII concentration in the normal population.Citation38 Six weekly replacements usually results in a serum FXIII level of 3%–5% towards the end of the period.Citation7
Approximately half of pregnant women with severe congenital FXIII deficiency can end up with spontaneous abortions and recurrent pregnancy losses, without proper replacement therapy.Citation80–Citation82 There is a scarcity of data on adequate replacement therapy for pregnancy. A plasma FXIII level of greater than 10% has been reported to be adequate to carry a pregnancy to term in deficient individuals.Citation83 Asahina et al reported that a replacement therapy with 250 IU per week was found to be sufficient to maintain a plasma level above 10%. The study recommended increasing the dose to 500 IU per week from the 23rd week of gestation to maintain pregnancy. A booster dose of 1000 IU can be given at the onset of labor to achieve plasma levels of over 30% to prevent severe hemorrhagic obstetric complications during labor.Citation83
Recently, ZymoGenetics Inc (Seattle, WA, USA) developed a novel recombinant FXIII-A2 (rFXIII-A2) that is free of human or mammalian products.Citation43 rFXIII-A combines with free endogenous FXIII-B in plasma to form a functional tetramer with a half-life of 10–14 days, similar to the endogenous form. However, in patients with congenital deficiency of FXIII-B, the plasma half-life of the functional tetramer becomes significantly shorter.Citation43 A phase 1 escalating dose trial conducted to evaluate the safety and efficacy of the novel recombinant rFXIII-A for long term prophylaxis concluded that the new product had a good safety profile, without any threatening adverse events or development of specific autoantibodies.Citation43 The study recommended monthly prophylaxis with rFXIII-A for the treatment of congenital FXIII deficiency. Inbal et al’s phase III trial of 2012 involving 41 patients with congenital FXIII deficiency reported five trauma-induced bleeding episodes in four patients treated with rFXIII-A2. Biochemical tests revealed transient, non-neutralizing, low volume antibody titers in the sera of four patients, but none of them developed any allergic or anaphylactic reactions, nor did they have any severe bleeding that required urgent intervention.Citation84 Although the antibodies developed in this study were clinically insignificant, in case of inhibitor development, the management typically involves immunosuppressive therapies including prednisone, cyclophosphamide, plasma exchange, intravenous immunoglobulin, and rituximab.Citation85
Conclusion
Congenital FXIII deficiency is a rare autosomal recessive disorder occurring in only one in three to five million people that remains underdiagnosed despite advances in the diagnostic modalities. Early recognition of this deficiency and adequate prophylactic therapy is crucial in order to save patients from life-threatening bleeding episodes and subsequent neurological morbidities and mortality. In this review, we have attempted to touch upon the recent advancements in the diagnosis and management of this congenital deficiency.
Although standard prophylaxis therapy includes FXIII concentrates, recombinant FXIII may be available in the future. Further studies are still needed to fully understand the varying functions of FXIII and how they impact clinical practice.
Disclosure
The authors report no conflicts of interest in this work.
References
- BoardPGLosowskyMSMiloszewskiKJFactor XIII: inherited and acquired deficiencyBlood Rev1993742292428130686
- LorandLLosowskyMSMiloszewskiKJHuman factor XIII: fibrin-stabilizing factorProg Hemost Thromb198052452907422874
- MiloszewskiKSheltawyMJLosowskyMSFibrinogen-fibrin degradation products and factor XIIIActa Haematol19745163213304212013
- OtakiMInabaHShinozawaKFujitaSAmanoKFukutakeKCharacterization of a large deletion that leads to congenital factor XIII deficiencyRinsho Byori2008563:187194 Japanese18411802
- FadooZSaleemAFFactor XIII deficiency in children – clinical presentation and outcomeJ Coll Physicians Surg Pak200818956556818803895
- PeyvandiFDugaSAkhavanSMannucciPMRare coagulation deficienciesHaemophilia20028330832112010428
- AnwarRMiloszewskiKJFactor XIII deficiencyBr J Haematol1999107346848410583246
- KosekiSSouriMKogaSTruncated mutant B subunit for factor XIII causes its deficiency due to impaired intracellular transportationBlood20019792667267211313256
- Gómez GarcíaEBPoortSRStibbeJTwo novel and one recurrent missense mutation in the factor XIII A gene in two Dutch patients with factor XIII deficiencyBr J Haematol2001112251351811167856
- AnwarRGallivanLTrinhCHillFMarkhamAIdentification of a new Leu354Pro mutation responsible for factor XIII deficiencyEur J Haematol200166213313611168522
- BhattacharyaMBiswasAAhmedRPClinico-hematologic profile of factor XIII-deficient patientsClin Appl Thromb Hemost200511447548016244775
- ShaikhANKhurshidMFactor XIII deficiency in PakistanJ Pak Med Assoc199343467698230654
- DuckertFJungEShmerlingDHA hitherto undescribed congenital haemorrhagic diathesis probably due to fibrin stabilizing factor deficiencyThromb Diath Haemorrh1960517918613724728
- MuszbekLBagolyZCairoAPeyvandiFNovel aspects of factor XIII deficiencyCurr Opin Hematol201118536637221738029
- KohlerHPIchinoseASeitzRAriensRAMuszbekLFactor XIII and fibrinogen SSC subcommittee of the ISTHDiagnosis and classification of factor XIII deficienciesJ Thromb Haemost2011971404140622946956
- SongJWChoiJRSongKSRheeJHPlasma factor XIII activity in patients with disseminated intravascular coagulationYonsei Med J200647219620016642548
- BoardPGWebbGCMcKeeJIchinoseALocalization of the coagulation factor XIII A subunit gene (F13A) to chromosome bands 6p24-p25Cytogenet Cell Genet198848125272903011
- YorifujiHAndersonKLynchGWVan De WaterLMcDonaghJB protein of factor XIII: differentiation between free B and complexed BBlood1988725164516503179443
- HsiehLNugentDFactor XIII deficiencyHaemophilia20081461190120019141159
- MuszbekLAdányRMikkolaHNovel aspects of blood coagulation factor XIII. I. Structure, distribution, activation, and functionCrit Rev Clin Lab Sci19963353574218922891
- WagnerBSeyfertUTGosseMWenzelEFickenscherKRuhlHGDetermination of factor XIII activity by a new photometric assay in plasma and platelets of healthy blood donorsThromb Res19947421691748029818
- KarimiMBereczkyZCohanNMuszbekLFactor XIII DeficiencySemin Thromb Hemost200935442643819598071
- KasaharaKSouriMKanedaMMikiTYamamotoNIchinoseAImpaired clot retraction in factor XIII A subunit-deficient miceBlood201011561277127919996413
- AndersenMDKjalkeMBangSCoagulation factor XIII variants with altered thrombin activation ratesBiol Chem2009390121279128319804366
- KobbervigCWilliamsEFXIII polymorphisms, fibrin clot structure and thrombotic riskBiophys Chem20041122–322322815572253
- AnwarRGallivanLRichardsMKhairKWrightMMinfordAFactor XIII deficiency: new nonsense and deletion mutations in the human factor XIIIA geneHaematologica200590121718172016330458
- IchinoseAPhysiopathology and regulation of factor XIIIThromb Haemost2001861576511487042
- LusherJPipeSWAlexanderSNugentDProphylactic therapy with Fibrogammin P is associated with a decreased incidence of bleeding episodes: a retrospective studyHaemophilia201016231632120017752
- LevyJHGreenbergCBiology of Factor XIII and clinical manifestations of Factor XIII deficiencyTransfusion Epub August 28, 2012
- IchinoseAHemorrhagic acquired factor XIII (13) deficiency and acquired hemorrhaphilia 13 revisitedSemin Thromb Hemost201137438238821805444
- LuoYYZhangGSAcquired factor XIII inhibitor: clinical features, treatment, fibrin structure and epitope determinationHaemophilia201117339339821323797
- MilnerGRHoltPJBottomleyJMaciverJEPractolol therapy associated with a systemic lupus erythematosus-like syndrome and an inhibitor to factor XIIIJ Clin Pathol1977308770773599192
- AhmadFSolymossSPoonMCBerubeCSullivanAKCharacterization of an acquired IgG inhibitor of coagulation factor XIII in a patient with systemic lupus erythematosusBr J Haematol19969337007038652397
- LorandLVelascoPTHillJMHoffmeisterKJKayeFJIntracranial hemorrhage in systemic lupus erythematosus associated with an autoantibody against actor XIIIThromb Haemost200288691992312529739
- KohlerHIchinoseASeitzRAriensRMuszbekLFactorXIII and fibrinogen SSC subcommittee of the ISTHDiagnosis and classification of factor XIII deficienciesJ Thromb Haemost2011971404140622946956
- LawrieASGreenLMackieIJLiesnerRMachinSJPeyvandiFFactor XIII – an under diagnosed deficiency – are we using the right assays?J Thromb Haemost20108112478248220727071
- IvaskeviciusVSeitzRKohlerHPStudy GroupInternational registry on factor XIII deficiency: a basis formed mostly on European dataThromb Haemost200797691492117549292
- AnwarRMinfordAGallivanLTrinhCHMarkhamAFDelayed umbilical bleeding – a presenting feature for factor XIII deficiency: clinical features, genetics, and managementPediatrics20021092E3211826242
- SchroederVDurrerDMeiliESchubigerGKohlerHPCongenital factor XIII deficiency in Switzerland: from the worldwide first case in 1960 to its molecular characterisation in 2005Swiss Med Wkly200713719–2027227817594539
- LakMPeyvandiFAli SharifianAKarimiKMannucciPMPattern of symptoms in 93 Iranian patients with severe factor XIII deficiencyJ Thromb Haemost2003181852185312911609
- MannucciPMFedericiABAntibodies to von Willebrand factor in von Willebrand diseaseAdv Exp Med Biol199538687928851017
- ToddTPerryDJA review of long-term prophylaxis in the rare inherited coagulation factor deficienciesHaemophilia200916456958319906159
- LovejoyAEReynoldsTCVisichJESafety and pharmacokinetics of recombinant factor XIII-A2 administration in patients with congenital factor XIII deficiencyBlood20061081576216556896
- VuralMYararCDurmazRAtasoyMASpontaneous acute subdural hematoma and chronic epidural hematoma in a child with F XIII deficiencyJ Emerg Med2010381252918514462
- MishraPNaithaniRDolaiTIntracranial haemorrhage in patients with congenital haemostatic defectsHaemophilia200814595295518637845
- CermeljMNegroFSchijmanEFerroAMAcerenzaMPollolaJNeurosurgical intervention in a haemophilic child with a subdural and intracerebral haematomaHaemophilia200410440540715230958
- SárváryASzucsSBaloghIPossible role of factor XIII subunit A in Fcgamma and complement receptor-mediated phagocytosisCell Immunol20042282819015219459
- NahrendorfMSosnovikDEWatermanPDual channel optical tomographic imaging of leukocyte recruitment and protease activity in the healing myocardial infarctCirc Res200710081218122517379832
- KappelmayerJBacskóGKelemenEAdányROnset and distribution of factor XIII-containing cells in the mesenchyme of chorionic villi during early phase of human placentationPlacenta19941566136237824447
- Koseki-KunoSYamakawaMDickneiteGIchinoseAFactor XIII A subunit-deficient mice developed severe uterine bleeding events and subsequent spontaneous miscarriagesBlood2003102134410441212933578
- ParameswaranKNChengXFChenECVelascoPTWilsonJHLorandLHydrolysis of gamma:epsilon isopeptides by cytosolic transglutaminases and by coagulation factor XIIIaJ Biol Chem19972721510311103179092583
- DargaudYde MazancourtPRugeriLAn unusual clinical presentation of factor XIII deficiency and issues relating to the monitoring of factor XIII replacement therapyBlood Coagul Fibrinolysis200819544745218600098
- LorandLGrayAJBrownKDissociation of the subunit structure of fibrin stabilizing factor during activation of the zymogenBiochem Biophys Res Commun19745649149224133182
- SchroederVKohlerHPNew developments in the area of factor XIIIJ Thromb Haemost201311223424423279671
- Al-JalladHFMyneniVDPiercy-KotbSAPlasma membrane factor XIIIA transglutaminase activity regulates osteoblast matrix secretion and deposition by affecting microtubule dynamicsPLoS One201161e1589321283799
- DardikRSolomonALoscalzoJNovel proangiogenic effect of factor XIII associated with suppression of thrombospondin 1 expressionArterioscler Thromb Vasc Biol20032381472147712805075
- DunnEJGrantPJType 2 diabetes: an atherothrombotic syndromeCurr Mol Med20055332333215892651
- KohlerHPInsulin resistance syndrome: interaction with coagulation and fibrinolysisSwiss Med Wkly200213219–2024125212148078
- IchinoseAFactor XIII is a key molecule at the intersection of coagulation and fibrinolysis as well as inflammation and infection controlInt J Hematol201295436237022477542
- LoofTGMorgelinMJohanssonLCoagulation, an ancestral serine protease cascade, exerts a novel function in early immune defenseBlood201111892589259821613262
- KissFSimonACsáthyLA coagulation factor becomes useful in the study of acute leukemias: studies with blood coagulation factor XIIICytometry A200873319420118000871
- KissFHevessyZVeszpremiALeukemic lymphoblasts, a novel expression site of coagulation factor XIII subunit AThromb Haemost200696217618216894461
- MiloszewskiKJALMIMJSeitzREgbringRFactor XIIISafety of long-term prophylaxis in inherited Factor XIII deficiencySecond International Conference1993Marburg, Stuttgart, GermanyFK Schattauer Verlagsgesellschaft mbH
- HussainRBittlesAHThe prevalence and demographic characteristics of consanguineous marriages in PakistanJ Biosoc Sci1998302:261275 http://www.ncbi.nlm.nih.gov/pubmed/?term=Hussain+R%2C+Bittles+AH.+The+prevalence+and+demographic+characteristics+of+consanguineous+marriages+in+Pakistan.+J+Biosoc+Sci.+1998%3B30(2)%3A261%E2%80%932759746828
- AntunesSVVicariPCavalheiroSBordinJOIntracranial haemorrhage among a population of haemophilic patients in BrazilHaemophilia20039557357714511296
- SørensenBYoungGGlobal Laboratory Assays in Hemophilia. Textbook of Hemophilia2nd ed2010263268
- SheltawyMJMiloszewskiKLosowskyMSFactors affecting factor XIII assay by dansyl cadaverine incorporationThromb Diath Haemorrh1972283:483488 http://www.ncbi.nlm.nih.gov/pubmed/46752694675269
- LorandLUrayamaTDe KiewietJWNosselHLDiagnostic and genetic studies on fibrin-stabilizing factor with a new assay based on amine incorporationJ Clin Invest1969486105410644977030
- LorandLCampbell-WilkesLKCoopersteinLA filter paper assay for transamidating enzymes using radioactive amine substratesAnal Biochem19725026236314674979
- LeeKNBirckbichlerPJPattersonMKJrColorimetric assay of blood coagulation factor XIII in plasmaClin Chem19883459069102897256
- WilmerMRudinKKoldeHEvaluation of a sensitive colorimetric FXIII incorporation assay. Effects of FXIII Val34 Leu, plasma fibrinogen concentration and congenital FXIII deficiencyThromb Res20011021819111323018
- MuszbekLPolgarJFesusLKinetic determination of blood coagulation Factor XIII in plasmaClin Chem198531135402856900
- FickenscherKAabAStüberWA photometric assay for blood coagulation factor XIIIThromb Haemost19916555355401871715
- AjznerEMuszbekLKinetic spectrophotometric factor XIII activity assays: the subtraction of plasma blank is not omissible [corrected]J Thromb Haemost20042112075207715550059
- DuckertFThe therapy of factor 13 deficiencyBibl Haematol196523135413575885230
- GootenbergJEFactor concentrates for the treatment of factor XIII deficiencyCurr Opin Hematol1998563723759814641
- CastamanGProphylaxis of bleeding episodes and surgical interventions in patients with rare inherited coagulation disordersBlood Transfus20086Suppl 2s39s4419105509
- HilgartnerMWMJGootenbergJEExperience with fibrogammin for the long term treatment of congenital factor XIII deficiency in the USABlood199178Suppl 160a
- NugentDJProphylaxis in rare coagulation disorders – factor XIII deficiencyThromb Res2006118Suppl 1S23S2816616323
- Bolton-MaggsPPerryDChalmersEThe rare coagulation disorders–review with guidelines for management from the United Kingdom Haemophilia Centre Doctors’ OrganisationHaemophilia2004105:593628 http://www.ncbi.nlm.nih.gov/pubmed/?term=The+rare+coagulation+disorders%E2%80%93review+with+guidelines+for+management+from+the+United+Kingdom+Haemophilia+Centre+Doctors%E2%80%99+Organisation15357789
- InbalAMuszbekLCoagulation factor deficiencies and pregnancy lossSemin Thromb Hemost200329217117412709920
- IchinoseAAsahinaTKobayashiTCongenital blood coagulation factor XIII deficiency and perinatal managementCurr Drug Targets20056554154916026274
- AsahinaTKobayashiTOkadaYGotoJTeraoTMaternal blood coagulation factor XIII is associated with the development of cytotrophoblastic shellPlacenta200021438839310833374
- InbalAOldenburgJCarcaoMRosholmATehranchiRNugentDRecombinant factor XIII: a safe and novel treatment for congenital factor XIII deficiencyBlood2012119225111511722451421
- AjznerESchlammadingerAKerényiASevere bleeding complications caused by an autoantibody against the B subunit of plasma factor XIII: a novel form of acquired factor XIII deficiencyBlood2009113372372518955560