
Abstract
The evolution of care in hemophilia is a remarkable story. Over the last 60 years, advances in protein purification, protein chemistry, donor screening, viral inactivation, gene sequencing, gene cloning, and recombinant protein production have dramatically enhanced the treatment and lives of patients with hemophilia. Recent efforts have produced enhanced half-life (EHL) clotting factors to better support prophylaxis and decrease the frequency of infusions. Medical needs remain in the areas of alternate modes of administration to decrease the need for venous access, better treatment, and prophylaxis for patients who form antibodies to clotting factors, and ultimately a cure of the underlying genetic defect. In this brief review, the authors summarize data on EHL clotting factors, introduce agents whose mode of action is not clotting factor replacement, and list current gene therapy efforts.
Introduction
Hemophilia is an inherited bleeding disorder. The most common forms, hemophilia A (one in every 5,000 live male births) and hemophilia B (one in every 30,000 live male births), are caused by the inheritance of abnormal forms of Factor VIII (FVIII) and Factor IX (FIX), respectively.Citation1,Citation2 Since the genes for both FVIII and FIX are located on the X chromosome, the disease displays the characteristic sex-linked pattern of inheritance. Since males have only one X chromosome, while females have two, males are most often affected. When a normal male and a female heterozygous for one abnormal FVIII (or IX) gene have children, male offspring have a 50% chance of inheriting hemophilia, and female offspring will have a 50% chance of carrying one abnormal gene copy. If a male with hemophilia has offspring with a female with two normal genes, all the males will be normal, and all the females will possess one copy of the abnormal gene. Due to the random shutdown of one of the pair of X chromosomes (the lyonization process), females may, if a higher percentage of the normal gene is silenced, have FVIII levels low enough to have increased bleeding. Recognized from biblical times, bleeding in affected males can occur virtually from birth. Thus, circumcision and heel sticks should be avoided in infants known to be at risk, and unsuspected occurrences may be diagnosed early due to excessive bleeding secondary to one of these procedures.
While greater than 300 unique mutations have been described for the FVIII gene, 40% of FVIII deficiency results from an inversion mutation of the short arm of the X chromosome, and a significant percentage are due to mutations of Xq28. While FIX deficiencies are also due to multiple mutations, the majority are due to mutations of Xq27.1-q27.2. The consequence of these mutations is a reduction in clotting factor activity. This might be due to production of a protein with decreased intrinsic activity, abnormal binding characteristics, or decreased plasma half-life. In this case, the antigenic amount of factor exceeds clotting factor activity. In other cases, very little of the abnormal protein is produced, and both factor antigen and activity are very low. In either case, the resultant decrease in activity puts the patient at risk of bleeding. Clotting factors are typically measured and reported as the number of units per given volume of plasma (1 unit/mL or 100 units/dL) or as a percent activity (100% = 100 units/dL). Since the amount of clotting factor varies significantly between individuals, a unit is defined as the amount of clotting factor activity in 1 mL of normal pooled plasma. Normal pooled plasma is prepared by pooling plasma from at least 20 normal volunteers. Given the marked variability of FVIII and FIX levels in normal individuals, a patient is considered to have a normal value if his level is greater than 50% but less than 150%. In hemophilia, the level of clotting factor activity is a primary determinant of the severity of the disease. Individuals with <1% activity have “severe” hemophilia with spontaneous bleeding.Citation3 Patients with factor levels between 1% and 5% have moderate disease with occasional spontaneous bleeding and severe bleeding with surgery or trauma. Individuals with factor levels >5% but <40% rarely have spontaneous bleeding but can bleed excessively bleed if challenged major surgery or trauma. While bleeding can occur at any location, hemorrhage into muscles or joints with little or no obvious trauma is the hallmark of hemophilia.
Evolution of modern hemophilia treatment
Prior to the 1960s, treatment of bleeding in hemophilia involved whole blood or plasma transfusions. In 1964, Judith Pool discovered that cryoprecipitate from plasma contains large amounts of FVIII.Citation4 This discovery led to treatment with cryoprecipitate followed by the development of lyophilized plasma concentrates of coagulation factors. These concentrates required donations from approximately 1,000 volunteers per vial. Their routine application in the 1970s improved the quality of life and extended the life expectancy of patients with hemophilia. Elective (even orthopedic) surgery became possible, and the availability of stable products enabled adoption of home treatment, facilitating early control of hemorrhage. While early treatment was good, it was demonstrated in Sweden that routine infusion of clotting factor in the non-bleeding state could actually prevent spontaneous bleeding and the severe joint damage that was caused by repetitive bleeding into joints.Citation5 This process of “primary prophylaxis” was rapidly adopted for younger patients with remarkable reductions in secondary arthropathy due to hemarthrosis.Citation6 Specialized hemophilia treatment centers came into existence, further enhancing and standardizing the care of hemophilia and providing routine access to orthopedic surgeons, physiotherapists, dentists, dieticians, and social workers. The discovery that 1-Desamino-8d-Arginine Vasopressin (DDAVP Desmopression Acetate) could be used to raise the level of FVIII in most patients with mild hemophilia A allowed the total avoidance of blood product exposure in these patients.Citation7 Within 15 years, the care of hemophilia patients dramatically improved.
Unfortunately, the promise of the 1970s suddenly darkened with the realization that the early plasma-derived concentrates were contaminated with life-threatening viral pathogens. Since bleeding episodes might require several vials of concentrates per event, patients with hemophilia were being exposed to blood from thousands of people with each treatment. While the risk of hepatitis from hepatitis B virus and the less well-characterized “Non-A Non-B” was known, the true depth of the danger was not appreciated until the emergence of the human immunodeficiency virus (HIV).Citation8 HIV and hepatitis C virus (HCV) were transmitted to patients with hemophilia. Virtually, all patients would develop hepatitis C with resultant risk of cirrhosis and hepatocarcinoma, and thousands would die in the 1980s and 1990s of AIDS.
This unforeseen disaster and the tragic consequences resulted in rapid technological and therapeutic advances. New ways and tests for screening donors were put into place. New methods to screen blood for viruses (nucleic acid amplification testing) were developed and implemented. Viral inactivation techniques (heat, solvent–detergent, nanofiltration, etc) were developed and incorporated as critical steps in the production of factor concentrates.Citation9 The genes for FVIII (1982) and FIX (1984) were cloned opening the way for the production of recombinant FVIII and FIX in the late 1980s and early 1990s.Citation10 Perhaps most importantly, the first effective antiviral treatments were developed allowing effective treatment of HIV and HCV. These advances set the stage for new and future treatments.
Current status
Clotting factor concentrates, both plasma derived and recombinant, have reached a very high level of purity. Nucleic acid testing testing (HIV 1-1, hepatitis B virus, HCV, hepatitis A virus, B19) and viral inactivation (dry heat, pasteurization, vapor, and solvent–detergent) for plasma-derived factor preparations have resulted in no blood-borne transmission of hepatitis or HIV in the last 25 years.Citation9 While having reached a high degree of safety, one documented case of parvovirus B19 transmission,Citation11 and the postmortem detection of variant Creutzfeldt–Jacob disease (vCJD) in a patient with hemophilia treated with plasma concentrates known to contain donations from a vCJD-infected donor, indicates that our guard cannot be dropped.Citation12 Therefore, removal of animal and human proteins from recombinant products has continued to advance with production of third-generation products devoid of albumin during production or formulation.
While the risk of infection has been minimized, the risk of antibody production against administered clotting factor continues to complicate the treatment or prevention of bleeding in patients who develop these “inhibitors”.Citation13 Inhibitors continue to be a major problem, and while treatment of acute bleeding in inhibitor patients is now possible in most cases, prophylaxis is poor, and immune tolerance induction is not uniformly effective.Citation14 Less immunogenic treatments and better ways to eradicate these antibodies are needed.
As a consequence of the advances outlined earlier, the life span of people with hemophilia is now similar to people without hemophilia, and we must pay increasing attention to the disorders of aging.
Remaining unmet medical needs
With the current hemophilia treatment paradigm, significant unmet need persists. While on-demand therapy for hemophilia A and B is very good with a variety of plasma-derived and recombinant factor concentrates, on-demand therapy for patients with inhibitors is not as reliable. While 80%–90% of these patients will respond to either recombinant activated Factor VIIa (rFVIIa) or Factor VIII inhibitor bypassing agent (FEIBA),Citation14 not all patients respond to both equally. Some patients may require a combination of rFVIIa and FEIBA which may be associated with an increased risk of thrombosis.Citation15,Citation16 However, several recent successful case reports and a small series of three patients treated safely with concomitant low doses of rFVIIa and FEIBA call into question the magnitude of the thrombotic risk.Citation17–Citation19
Prophylactic treatment for hemophilia B is good with the majority of patients having virtual elimination of spontaneous bleeding with one or two infusions per week. With the introduction of extended half-life FIX molecules, prophylactic therapy for hemophilia B promises to be excellent with even further reductions in dosing frequency. Prophylaxis in patients with hemophilia A generally requires two to three infusions per week, and the enhanced half-life (EHL) FVIII agents may not adequately support once-per-week prophylaxis in all patients. Prophylaxis in inhibitor patients is especially problematic with only one agent, FEIBA, having the approved indication and therapy requiring rather large volume (75 mL) intravenous (IV) infusions of product every other day to provide a 60% reduction in spontaneous bleeding.Citation20 Alternate modes of administration of treatment (subcutaneous [SC] or even oral) are needed, and innovative nonfactor replacement therapies may offer broader spectrum treatments. At the present time, cures for hemophilia A and B are not available. While it is clear that stopping bleeding is good and preventing bleeding is better, cure remains the ultimate goal in the quest to normalize the lives of patients with hemophilia.
In the following sections, we briefly review the new extended half-life clotting factor products, newer agents being developed for patients with inhibitors, and new nonfactor replacement therapies for hemophilia. We conclude with a brief summary of current gene therapy efforts.
Extended half-life clotting factor products
rFIX:Fc
Recent advances in hemophilia treatment have centered on linkage of native, recombinant FIX (rFIX) to various molecules designed to prolong the intravascular residence of the molecule. These efforts have utilized the common, current strategies employed to extend the half-life of protein therapeutics: linkage to the fragment crystallizable (Fc) region of human antibodies, to polyethylene glycols (PEGs) of various sizes (PEGylation), and to recombinant albumin ().
Table 1 New factor EHL-FVIII agents
A single molecule of rFIX is fused covalently and directly without linkers to the dimeric Fc of IgG1 (rFIX:Fc) and has been named “eftrenonacog alfa”. It is currently approved to use in the USA as Coagulation Factor IX (Recombinant), Fc Fusion Protein (ALPROIX, Biogen Idec, Cambridge, MA, USA). The molecule is expressed in HEK-293H cells, a human cell line in a serum-free medium.Citation21 Linkage to the Fc prolongs the intravascular residence and hence biologic action of FIX by utilizing the Fc intracellular trafficking process. There may also be a component of reduced renal clearance.Citation22
The Fc trafficking process utilizes the body’s pathway whereby proteins are endocytosed and sorted. Non-bound endosome proteins are sorted to intracellular degradation in lysosomes, while bound proteins (eg, Fc bound to the neonatal Fc-receptor) are routed for exocytosis.Citation23,Citation24 While trafficking through the endosome sorting/exocytosis processes, the FIX is shielded from intravascular degradation proteases.Citation25 Initial trials yielded favorable safety results.Citation26,Citation27 FIX:Fc is denominated in FIX international units (IU) where 1 IU is defined as the amount of FIX in 1 mL of normal pooled plasma, and dosing performed on the basis of a patient’s weight in kilogram. Fc linkage does not affect coagulation factor activity but does confer an extended half-life (geometric mean =82.1 hours) compared to native FIX (nonacog alfa, geometric mean =33.8 hours) using a 96-hour sampling schedule. The incremental recovery of FIX:Fc (geometric mean =0.92, 95% confidence interval [CI] 0.77–1.10) was comparable to standard FIX (nonacog alfa, geometric mean =0.95, 95% CI 0.81–1.01).Citation28 The model-predicted percentage of patients with a trough level >1 IU/dL and >3 IU/dL after weekly dosing was higher with FIX:Fc compared to native FIX (nonacog alfa; ).Citation29
Table 2 Predicted proportion of patients with FIX trough levels above 1 IU/dL and 3 IU/dL (steady state after six doses) with weekly dosing
In the pivotal trial, annualized bleeding rates (ABRs) of prophylaxis regimens once weekly (Group 1) or initially once every 10 days (Group 2, interval adjusted as needed) were compared with on-demand therapy (Group 3, episodic). Low ABRs were achieved in both prophylaxis regimens (), and overall, 23% of patients receiving weekly dosing and 42.3% of patients receiving adjusted interval dosing experienced no bleedings during the trial.Citation28 Efficacy of acute bleed treatment was 90.4% with a single dose and 97.3% with one or two doses. No inhibitors or vascular thrombotic events were detected. Adverse events were noted in 73.9% of patients, and 10.9% had at least one serious adverse event with only one event judged possibly related to rFIX:Fc, an obstructive ureteral clot which resolved with medical management.Citation30
Table 3 Annualized bleeding rates by treatment regimen, rFIX:Fc
When rFIX:Fc was used during surgery, only a single dose was needed to maintain hemostasis in 85.7% of patients (presurgery to the end of the operation). Two to three infusions were required on postoperative days 1–3, and no patient required dosing every day during the perioperative interval (days 0–14). One dose was required in most minor surgery cases. All major and 83% of minor surgeries reported excellent/good hemostasis. Two out of 12 patients with major surgeries required transfusions (one patient with three packed red blood cell units and one patient with seven total units of packed red blood cells and fresh frozen plasma, individual breakdown not reported). Adverse events were reported in 83.3% of major surgeries (six serious adverse events), and none of the adverse events were related to rFIX:Fc. No patient developed inhibitors, had an anaphylactic reaction, or experienced a vascular thrombotic event.Citation30
FIX:PEG
Conjugation of proteins to PEG has been employed to prolong their duration of action. Controlled PEGylation is employed, since random PEGylation tended to increase protein immunogenicity.Citation31,Citation32 Targeted PEGylation is important so as to preserve the catalytic activity of the protein while achieving a prolonged duration of action.
GlycoPEGylated rFIX (rFIX:PEG), nonacog beta pegol, is currently in clinical trials. rFIX is expressed in Chinese hamster ovary cells, and glycoPEGylation (40 kDa) is performed at the activation peptide, so the PEG is jettisoned upon FIX activation.Citation33 When FIX is activated, two internal peptide bonds are broken (specific arginine–alanine and arginine–valine bonds) releasing a small “activation peptide” with the resulting larger protein now termed “activated FIX (FIXa)”. Since the PEG moiety is linked to the activation peptide, the PEG is jettisoned along with the activation peptide when FIX is activated. PEGylation within the activation peptide domain was shown to maintain catalytic activity and prolong half-life approximately fivefold.Citation34,Citation35 The Phase I trial showed IV tolerability with one hypersensitivity reaction attributed to the molecule.Citation35
A single IV dose and steady-state pharmacokinetic (PK) study showed a mean half-life of 85 hours (coefficient of variation percent [CV%] =21.8) in the 10 IU/kg group and 111 hours (CV% =11.8) in the 40 IU/kg group. The estimated mean 7-day rFIX trough levels after IV injection were 8.5 IU/dL (95% CI 7.7–9.3) for the 10 IU/kg group and 27.3 IU/dL (95% CI 24.8–30.0) for the 40 IU/kg group.Citation36
In the Phase III pivotal trial, rFIX:PEG weekly IV prophylaxis at 10 IU/kg or 40 IU/kg was compared with IV on-demand dosing. Both prophylaxis arms had lower ABRs than on-demand with the 40 IU/kg having the lowest ABR (). During the prophylaxis period, 45% of patients in the 40 IU/kg group and 17% of those in the 10 IU/kg group experienced no treatment-requiring bleeding episodes. Target joint bleeding resolution was achieved in 66.7% in the 40 IU/kg group and in 7.7% in the 10 IU/kg group. When treating bleeding episodes, in the 40 IU/kg group, 99% resolved with one dose, while in the 10 IU/kg and on-demand groups, 84% resolved with one dose. No inhibitors developed, and no deaths or thromboembolic events were noted. Adverse events were experienced by 81% of patients (3.33 adverse events per patient-year), and the serious adverse event rate was 5.4% with all being unrelated to rFIX:PEG.Citation36
Table 4 Annualized bleeding rates by treatment regimen, rFIX:PEG
rFIX:Albumin
Albumin is the most plentiful plasma protein with a half-life of 20 days. Fusion to albumin prolongs drug half-life via the intracellular processing/trafficking process as well as reduced renal clearance for large molecules.Citation22 Recombinant albumin is also well characterized. rFIX:Albumin is produced in Chinese hamster ovary cells with albumin linked to the C-terminus of rFIX with a cleavable linker derived from region Ser136 or Val137 to Val153 covering the cleavage site composed of the C-terminus of the FIX light chain which was designed to allow albumin removal in parallel with FIX activation.Citation37 This linkage differs from the FIX:PEG construct (activation peptide linkage) but accomplishes the same goal which is to release the albumin from FIXa so as to neither affect the activity of FIX once activated nor prolong the activity of FIXa once formed.Citation37
A Phase I dose escalation study showed that rFIX:albumin was well tolerated with no hypersensitivity reactions noted. The mean (standard deviation) half-life was assessed over three doses and was 104.71 (55.08) hours for 25 IU/kg, 91.57 (20.74) hours for 50 IU/kg, and 98.82 (17.48) hours for 75 IU/kg. The mean (standard deviation) FIX activity on 168 hours (7 days) after a single dose of 25 IU/kg was 7.41 (3.87) IU/dL, for 50 IU/kg was 13.41 (2.91) IU/dL, and for 75 IU/kg was 17.39 (4.46) IU/dL.Citation38,Citation39
New agents for patients with inhibitors
OBIZUR
Due to variations in the amino acid sequence between human and porcine FVIII, there is reduced reactivity of human FVIII antibodies to porcine FVIII. As a consequence, porcine FVIII can be used to treat bleeding episodes in patients with human FVIII antibodies with no or low cross reactivity with porcine FVIII. Plasma-derived porcine FVIII was utilized for the treatment of bleeding in such patients, until it was withdrawn from the market in 2004. Withdrawal of Hyate C was due to the detection of low levels of parvovirus in the drug product. Subsequently, a recombinant B-domain truncated porcine FVIII (OBI-1) containing a 24-amino acid linker comprising the first and last 12 amino acids of the B-domain was developed. First-in-human trials of this molecule revealed higher bioavailability than Hyate C in hemophilia A patients without antibodies to porcine FVIII, and the preparation was found to be well tolerated.Citation40 Subsequent development and clinical trials led, in 2014, to the approval of OBI-1 (OBIZUR) for treatment of bleeding episodes in patients with acquired hemophilia A.Citation41 The 29 enrolled patients who spontaneously developed antibodies to FVIII had a variety of underlying conditions including malignancies and autoimmune disorders. Ten out of the 29 patients had baseline anti-porcine FVIII cross reactivity, and one failed treatment inclusion criteria. Control of qualifying bleed was ultimately achieved in 24 out of 28 patients. Approval for treatment in hemophilia A patients who develop inhibitors has not been sought to this point ().
Table 5 New agents for hemophilia patients with inhibitors
Alb-rFVIIa-FP
Alb-rFVIIa-FP is a fusion protein (FP) linking human coagulation Factor VIIa (FVIIa) and human albumin. A single recombinant gene construct is expressed in Chinese hamster ovary cells yielding a recombinant human albumin fused to the C-terminus of rFVIIa via a flexible glycine–serine linker.Citation42–Citation44 Preclinical studies in hemophilia A mice, rats, rabbits, and cynomolgus monkeys confirmed variable but consistent prolongations of the PK effects of Alb-rFVIIa-FP versus rFVIIa.Citation38 PK half-life was prolonged four to five times that of rFVIIa in the various animals. The potency of rFVIIa was noted to be 2.7-fold higher than rFVIIa-FP. This molecule is now in clinical trials, and with a half-life of 8.5 hours, holds promise for on-demand and prophylaxis treatment in hemophilia patients with inhibitors.
EHL-rFVIIa-CTP
FVIIa-CTP (mod-5014), a novel long-acting coagulation factor, displays a prolonged hemostatic effect following IV and SC administration in hemophilic animal models.Citation45 When PK parameters for rFVIIa and FVIIa-CTP were compared in animal models, the half-life and area under the curve of FVIIa-CTP were five- and threefold higher. The carboxyl terminal peptide addition is small and does not significantly increase the solution viscosity. IV and SC administration in animals confirmed the potential for SC administration. Development of the molecule continues with human trials planned to evaluate safety and efficacy of both IV and SC administration in hemophilia patients with inhibitors.
TheraPEG-Factor VIIa
TheraPEG™ is a conjugation technology for site-specific PEGylation at solvent-accessible disulfide bonds.Citation46 PolyTherics Ltd is applying this technique to conjugate PEG to recombinant human clotting factors at a site remote from the active site in an effort to cover epitopes responsible for eliciting an immune response. The goal is to retain clotting activity while reducing immunogenicity, prolonging the plasma half-life and allowing small-volume SC administration of clotting factors. TheraPEG-F VIIa has been administered to hemophilic dogs by both IV and SC routes. When given intravenously, the time over which the whole blood clotting time remained less than 12 minutes (96 hours) was 12 times longer than that seen with rFVIIa (8 hours). When administered via the SC route, TheraPEG-F VIIa had 89% bioavailability and maintained the whole blood clotting time to less than 12 minutes for 72 hours. In the same dog model, rFVIIa given subcutaneously at an equivalent dose did not correct the whole blood clotting time and continues in development.
rFVIIaFc
Recombinant FVIIaFc (rFVIIaFc) is a recombinant Fc FP which uses the neonatal Fc receptor-mediated recycling pathway to protect the FP from catabolism.Citation47 Based on activity, rFVIIaFc has a 5.5 times longer terminal half-life than rFVIIa in hemophilic mice. In a thrombin generation assay and in hemophilia A mouse tail clip model, the potency of rFVIIaFc was comparable to rFVIIa at comparable molar doses. The potential of rFVIIaFc to enable prolonged efficacy awaits additional investigational confirmation.
Nonfactor replacement therapies for hemophilia
Recently, several groups have departed from the normal paradigm of developing new and better versions of replacement clotting factors, to explore the potential and ability of non-clotting factor replacements to correct hemostasis in patients with hemophilia. On the one hand, several approaches involve decreasing the capacity of normal clotting factor inhibitors (tissue factor pathway inhibitor [TFPI], antithrombin, etc) to tamp down clotting activity. Another approach is to engineer a completely new protein to mimic or replace the activity of a protein that is either missing from birth or has been removed by an antibody. While all are relatively early in their development, all have reached human trials and appear to hold promise for treatment across a variety of hemophilia patients. These approaches are summarized in .
Table 6 Nonfactor replacement therapies
NN-7415 (concizumab/mAB2021) (anti-TFPI)
Concizumab is an anti-TFPI antibody that binds to the K2 domain of TFPI.Citation48 The goal is to reduce the inhibitor activity of TFPI against the tissue factor pathway and thereby enhance the ability of this pathway to support hemostasis. By so doing, the need for intrinsic pathway support of activated Factor X (FXa) production is reduced. As a consequence, the amount of FVIII and FIX needed for normal function is decreased. Since this mechanism should be relevant to FIX deficiency, FVIII deficiency, and to patients with inhibitors to these proteins, this approach may have broader potential applications. The safety and PKs of this molecule have been studied in healthy volunteers and in patients with hemophilia.Citation49 In a first-in-human, Phase I, multicenter, randomized, double-blind, placebo-controlled trial, escalating single IV (0.5–9,000 µg/kg) or SC (50–3,000 µg/kg) doses of concizumab were given to healthy volunteers (n=28) or hemophilia patients (n=24). No serious adverse events occurred, and no antidrug antibodies were seen. Dose- dependent procoagulant effects were noted as increased D-dimers and prothrombin 1 + 2 levels. Noted nonlinear PKs was felt to be due to target-mediated clearance. Further studies are planned to evaluate the potential of SC-administered concizumab in hemophilia treatment.
ALN-AT3 (Alnylam)
ALN-AT3 is an RNAi therapeutic targeting antithrombin 3 (AT3). In a manner analogous to anti-TFPI, ALN-AT3 seeks to reduce the requirement for clotting factors required for the production of FXa by reducing the inhibition of FXa by AT3. Since AT3 is also a primary controller of FXI, FIX, FII, and to some degree FVII, this approach has the potential to modify a variety of clotting factor deficiencies. In animal models, ALN-AT3 yields potent (up to 100%), dose-dependent (1–30 mg/kg), and durable (>30 days) knockdown of AT3.Citation50 This silencing of antithrombin results in up to a fourfold increase in thrombin generation. In a Phase I multiple ascending SC dosing study in normal volunteers (low dose only) and patients with hemophilia A or B, 70% AT3 knockdown with concurrent 334% increase in thrombin generation was noted at a 45 µg/kg dose given weekly for 3 weeks. The AT3 nadir occurred at 28 days. As with the animal models, the effect was durable up to 60 days at the 15 µg/kg dose.Citation51 Further studies are planned to evaluate effects at longer dosing intervals.
ACD-910
ACD-910 is a bispecific antibody with binding specificity for FIX, FIXa, FX, and FXa.Citation52–Citation56 It is a humanized, monoclonal modified antibody designed to replace the cofactor function of FVIII by bringing FIXa and FX into approximate orientation for rapid conversion of FX to FXa. The mode of action is nonclotting factor replacement of FVIII activity, thereby reducing or eliminating the need for FVIII. In a Phase I trial of three dose levels (1 mg/kg loading followed by 0.3 mg/kg weekly, 3 mg/kg loading followed by 1 mg/kg weekly, and 3 mg/kg weekly) given SC, the mean half-life of the molecule was noted to be 28–34 days. Greater than 5% FVIII activity levels were achieved with marked reductions in bleeding even at the lowest dose. At the highest dose, no bleeding occurred. In this 12-week study, anti-ACE-910 antibody developed in one patient. No significant safety concerns have been identified, and additional trials are planned to evaluate the safety and efficacy of this agent for prophylaxis in patients with hemophilia A with and without inhibitors.
Gene therapy for hemophilia
While a review of current activity in gene therapy for hemophilia is beyond the scope of this short review, it is instructive to consider the magnitude of the effort summarized in . Multiple universities, biotechnology enterprises, and most recently larger pharmaceutical companies have coalesced around the development of gene therapy for hemophilia. After the missteps of the late 1990s, steady progress has led to the current state. Apparently, safe adeno-associated viral (AAV) vectors have been identified for delivery of the FIX gene to hematocicytes in a manner similar to that illustrated for adenovirus in . FIX activity levels, sufficient to dramatically reduce spontaneous bleeding, have been obtained in patients with severe disease, and these levels have been maintained for greater than 4 years in some patients.Citation57–Citation59 The use of high-activity variants of FIX may allow levels of activity approaching normal to be achieved without additional improvements in the levels of transduction.Citation60 The importance of preexisting antibodies to vectors and the occurrence of cell-specific T-cell responses capable of eradicating transduced cells have been identified, and approaches to avoidance and treatment are being developed.
Figure 1 Gene therapy using an adenovirus vector.
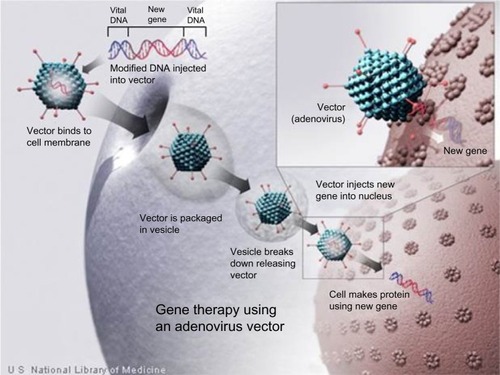
Table 7 Gene therapy programs for hemophilia
The progress in gene therapy for hemophilia A has been hampered to a significant degree by the size of the gene. While the FIX gene is accommodated nicely in AAV vectors, the FVIII gene barely fits. While modifications to the FVIII gene cassette to allow accommodation in AAV vectors may offer one avenue to solve this problem, advances in the use of lentiviral vectors that are capable of carrying the FVIII gene may also provide the solution.
Lentiviral vectors potentially offer significant advantages. They are able to efficiently integrate into the target genome, even when the cells are not actively dividing.Citation61 If the liver is the target organ, this can lead to transduction of nondividing hepatocytes with the potential of long-term expression. The liver also contains an abundance of antigen-presenting cells (APCs) that can also be transduced. Transduction of APCs might increase the risk of an immune response to FVIII or FIX and thereby inhibit long-term expression.Citation62 The use of hepatocyte-specific promoters and incorporation of hematopoietic-specific microRNA target sequences into the vector many reduce this immune risk.Citation63–Citation66 While the ability to incorporate endows long-term expression, it also raises concerns regarding insertional mutagenesis caused by oncogene activation or inactivation of tumor-suppressor genes.Citation61 This risk may be reduced by partial removal of the long terminal repeat or by the introduction of an inactivating mutation in the integrase to produce an integration-defective lentiviral vector.Citation61,Citation66 Progress in this area continues at a rapid pace and holds promise to solve the problem of FVIII gene delivery.
Gene therapy for hemophilia B is on the horizon, and gene therapy for hemophilia A is becoming feasible. The question could be asked – why is this being pursued when long-acting agents for hemophilia B should reduce the burden of prophylaxis and some of the agents discussed earlier hold promise to do the same for hemophilia A? The answer is probably found in the fact that while all these advances are improving the lives of patients with hemophilia, all will still require recurrent chronic treatment. The majority of these treatments will require a needle either in a vein or in the skin. There is a desire on the part of patients to be free of these treatments and to be free of the risk of bleeding. A desire to be normal drives the quest for a cure. If stopping bleeding is good, and preventing bleeding is better, then curing the disease must be the best treatment goal.
Disclosure
The Authors are employees of Pfizer Inc. This information represents the authors’ views and does not necessarily reflect those of Pfizer. The authors report no other conflicts of interest in this work.
References
- MannucciPMTuddenhamEGDThe hemophiliac – from royal genes to gene therapyN Engl J Med20013441773177911396445
- Bolton-MaggsPHPasiKJHemophilias A and BLancet20033611801180912781551
- WhiteGC2ndRosendaalFAledortLMLusherJMRothschildCIngerslevJDefinitions in hemophilia. Recommendation of the scientific subcommittee on factor VIII and factor IX of the scientific and standardization committee of the International Society on Thrombosis and HaemostasisThromb Haemost20018556011307831
- MannucciPMHemophilia and related bleeding disorders: a story of dismay and successHematology Am Soc Hematol Educ Program200211912446416
- NilssonIMExperience with prophylaxis in SwedenSemin Hematol1993303 Suppl 216198367738
- Manco-JohnsonMJAbshireTCShapiroADProphylaxis versus episodic treatment to prevent joint disease in boys with severe hemophiliaN Engl J Med200735753554417687129
- MannucciPMDesmopressin (DDAVP) in the treatment of bleeding disorders: the first twenty yearsHaemophilia20006Suppl 1606710982270
- MannucciPMHemophilia: treatment options on the twenty-first centuryJ Thromb Haemost200511349135512871268
- MannucciPMBack to the future: a recent history of haemophilia treatmentHaemophilia200814Suppl 3101818510516
- WhiteGCMcMillanCWKingdonHSShoemakerCBUse of recombinant antihemophilic factor in the treatment of two patients with classic hemophiliaN Engl J Med19893201661702492083
- SoucieJMDe StaerckeCMonahanPEUS Hemophilia Treatment Center Network. Evidence for the transmission of parvovirus B19 in patients with bleeding disorders treated with plasma-derived factor concentrates in the era of nucleic acid test screeningTransfusion2013531217122522998193
- LlewelynCAHewittPEKnightRSPossible transmission of variant Creutzfeldt-Jakob disease by blood transfusionLancet2004363940741742114962520
- AstermarkJOverview of inhibitorsSemin Hematol2006432 Suppl 4S3S716690373
- AstermarkJMoradoMRocinoAEHTSBCurrent European practice in immune tolerance induction therapy in patients with haemophilia and inhibitorsHaemophilia20061236337116834735
- NgHJLohYSMTanDCLLeeLHThrombosis associated with the use of recombinant activated factor VII: profiling two eventsThromb Haemost2004921448144915624250
- RosenfeldSBWatkinsonKKThompsonBHMacfarlanDELentzSRPulmonary embolism after sequential use of recombinant factor VIIa and activated prothrombin complex concentrate in a factor VIII inhibitor patientThromb Haemost20028792592612038803
- EconomouMTeliATzantzaroudiATsatraIZavitsanakisAAthanassiou-MetaxaMSequentila therapy with activated prothrombin complex concentrate (FEIBA) and recombinant factor VIIa in a patient with severe haemophilia A, inhibitor presence and refractory bleedingHaemophilia20081439039118194305
- MirandGGRodgersGMTreatment of an acquired factor VIII inhibitor with sequential recombinant factor VIIa and FEIBAHaemophila200915383385
- MartinowitzULivnatTZivelinAKenetGConcomitant infusion of low doses of rFVIIa and FEIBA in haemophilia patients with inhibitorsHaemophilia20091590491019473416
- AntunesSVTangadaSStasyshynORandomized comparison of prophylaxis and on-demand regimens with FEIBA NF in the treatment of haemophilia A and B with inhibitorsHaemophilia201420657223910578
- DucoreJMMiguelinoMGPowellJSAlprolix (recombinant factor IX Fc fusion protein): extended half-life product for the prophylaxis and treatment of hemophilia BExpert Rev Hematol2014755957125142322
- KontermannREStrategies for extended serum half-life of protein therapeuticsCurr Opin Biotechnol20112286887621862310
- GoeblNABabbeyCMDatta-MannanAWitcherDRWroblewskiVJDunnKWNeonatal Fc receptor mediates internalization of Fc in transfected human endothelial cellsMol Biol Cell2008195490550518843053
- RathTBakerKDumontJAFc-fusion proteins and FcRn: structural insights for longer-lasting and more effective therapeuticsCrit Rev Biotechnol20153523525424156398
- TakakiAEnfieldDLThompsonARCleavage and inactivation of factor IX by granulocyte elastaseJ Clin Invest198372170617156605369
- ShapiroADRagniMVValentinoLARecombinant factor IX-Fc fusion protein (rFIXFc) demonstrates safety and prolonged activity in a phase 1/2a study in hemophilia B patientsBlood201211966667222110246
- PetersRTLowSCKamphausGDProlonged activity of factor IX as a monomeric Fc fusion proteinBlood20101152057206420056791
- PowellJSPasiKJRagniMVB-LONG InvestigatorsPhase 3 study of recombinant factor IX Fc fusion protein in hemophilia BN Engl J Med20133692313232324304002
- PowellJSShapiroARagniMSwitching to recombinant factor IX Fc fusion protein prophylaxis results in fewer infusions, decreased factor IX consumption and lower bleeding ratesBr J Haematol201516811312325209873
- PowellJSApteSChambostHLong-acting recombinant factor IX Fc fusion protein (rFIXFc) for perioperative management of subjects with haemophilia B in the phase 3 B-LONG studyBr J Haematol201516812413425208598
- AbuchowskiAvan EsTPalczukNCDavisFFAlteration of immunological properties of bovine serum albumin by covalent attachment of polyethylene glycolJ Biol Chem197725235783581405385
- MeiBPanCJiangHRational design of a fully active, long-acting PEGylated factor VIII for hemophilia A treatmentBlood201011627027920194895
- CollinsPWMøssJKnobeKGrothAColbergTWatsonEPopulation pharmacokinetic modeling for dose setting of nonacog beta pegol (N9-GP), a glycoPEGylated recombinant factor IXJ Thromb Haemost2012102305231222998153
- ØstergaardHBjelkeJRHansenLProlonged half-life and preserved enzymatic properties of factor IX selectively PEGylated on native N-glycans in the activation peptideBlood20111182333234121700771
- NegrierCKnobeKTiedeAGiangrandePMossJEnhanced pharmacokinetic properties of a glycoPEGylated recombinant factor IX: a first human dose trial in patients with hemophilia BBlood20111182695270121555744
- CollinsPWYoungGKnobeKParadigm 2 InvestigatorsRecombinant long-acting glycoPEGylated factor IX in hemophilia B: a multinational randomized phase 3 trialBlood20141243880388625261199
- MetznerHJWeimerTKronthalerULangWSchulteSGenetic fusion to albumin improves the pharmacokinetic properties of factor IXThromb Haemost200910263464419806248
- SantagostinoENegrierCKlamrothRSafety and pharmacokinetics of a novel recombinant fusion protein linking coagulation factor IX with albumin (rIX-FP) in hemophilia B patientsBlood20121202405241122859609
- SantagostinoEPROLONG-9FP clinical development program – phase I results of recombinant fusion protein linking coagulation factor IX with recombinant albumin (rIX-FP)Thromb Res2013131Suppl 2S7S1023537724
- KemptonCLAbshireTCDeverasRAPharmacokinetics and safety of OBI-1, a recombinant B domain-deleted porcine factor VIII, in subjects with haemophilia AHaemophilia20121879880422512291
- Jruse-JarresRSt-LouisJGreistAEfficacy and safety of OBI-1, an antihaemophilic factor VIII (recombinant), porcine sequence, in subjects with acquired haemophilia AHaemophilia20152116217025623166
- ZollnerSSchuermannDRaquetEPharmacological characteristics of a novel, recombinant fusion protein linking coagulation factor VIIa with albumin (rFVIIa-FP)J Thromb Haemost20141222022824641308
- SchulteSUse of albumin fusion technology to prolong the half-life of recombinant factor VIIaThromb Res2008122Suppl 4S14S1918929521
- WeimerTWormsbächerWKronthalerULangWLiebingUSchulteSProlonged in-vivo half of factor VIIa by fusion to albuminThromb Haemost20089965966718392323
- HartGHershkovitzOLilanABZakarMBinderLFimaEMod-5014, a novel long-acting FVIIa proposing an improved prophylactic and on demand treatment for hemophilia patients following SC and IV administration comprehensive in-vitro and in-vivo evaluation in preparation for clinical studiesBlood20131223578
- BalanSChoiJWGodwinASite-specific PEGylation of protein disulfide bonds using a three-carbon bridgeBioconjug Chem200718617617226958
- SalasJLiuTLuQEnhanced pharmacokinetics of factor VIIa as a monomeric Fc fusionThromb Res201513597097625721936
- AgersøHOvergaardRVPetersenMBPharmacokinetics of an anti-TFPI monoclonal antibody (concizumab) blocking the TFPI interaction with the active site of FXa in cynomolgus monkeys after iv and sc administrationEur J Pharm Sci201456656924568891
- ChowdaryPLethagenSFriedrichUThe Explorerâ1 InvestigatorsSafety and pharmacokinetics of anti-TFPI antibody (concizumab) in healthy volunteers and patients with hemophilia: a randomized first human dose trialJ Thromb Haemost20151374375425641556
- BarrosSACariotoMHettingerJExpanded therapeutic index of antithrombin silencing and correction of APTT in a hemophilia A mouse modelBlood20131223585
- SorensenBMantTAkincAAln-AT3 Investigators, International Multicenter StudyA subcutaneously administered RNAi therapeutic (ALN-AT3) targeting antithrombin for treatment of hemophilia: interim phase 1 study results in healthy volunteers and patients with hemophilia A or BBlood2014124693
- SampeiZIgawaTSoedaTNon-antigen-contacting region of an asymmetric bispecific antibody to factors IXa/X significantly affects factor VII-mimetic activityMAbs2015712012825524207
- KitazawaTIgawaTSampeiZA bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A modelNat Med2012181570157423023498
- SampeiZIgawaTSoedaTIdentification and multidimensional optimization of an asymmetric bispecific IgG antibody mimicking the function of factor VIII cofactor activityPLoS One20138e5747923468998
- MutoAYoshihashiKTakedaMAnti-factor IXa/X bispecific antibody (ACE910): hemostatic potency against ongoing bleeds in a hemophilia A model and the possibility of routine supplementationJ Thromb Haemost201412206213
- MutoAYoshihashiKTakedaMThe anti-factor IXa/X bispecific antibody ACE910 prevents spontaneous joint bleeds in a long-term primate model of acquired hemophilia ABlood20141243165317125274508
- HighKHNathwaniASpencerTLillicrapDCurrent status of haemophilia gene therapyHaemophilia201420Suppl 4434924762274
- NathwaniACTuddenhamEGRangarajanSAdenovirus-associated virus vector-mediated gene transfer in hemophilia BN Engl J Med20113652357236522149959
- NathwaniACReissUMTuddenhamEGLong-term safety and efficacy of factor IX gene therapy in hemophilia BN Engl J Med20143711994200425409372
- CrudeleJMFinnJDSinerJIAAV liver expression of FIX-Padua prevents and eradicates FIX inhibitor without increasing thrombogenicity in hemophilia B dogs and miceBlood20151251553156125568350
- MatraiJChuahMKVandenDriesscheTRecent advance in lentiviral vector development and applicationsMol Ther20101847749020087315
- VandenDriesscheTThorrezLNaldiniLLentiviral vectors containing the human immunodeficiency virus type-1 central polypurine tract can efficiently transduce nondividing hepatocytes and antigen-presenting cells in vivoBlood200210081382212130491
- FollenziABattagliaMLombardoAAnnoniARoncaroloMGNaldiniLTargeting lentiviral vector expression to hepatocytes limits transgene-specific immune response and establishes long-term expression of human antihemophilic factor IX in miceBlood20041033700370914701690
- VandenDriesscheTThorrezLAcosta-SanchezAEfficacy and safety of adeno-associated viral vectors based on serotype 8 and 9 vs lentiviral vectors for hemophilia B gene therapyJ Thromb Haemost20075162417002653
- BrownBDCantoreAAnnoniAA microRNA-regulated lentiviral vector mediates stable correction of hemophilia B miceBlood20071104144415217726165
- MatraiJCantoreABartholomaeCCHepatocyte-targeted expression by integrase-defective lentivrial vectors induces antigen-specific tolerance in mice with low genotoxic riskHepatology2011531696170721520180
- US National Library of MedicineGene therapy using an adenovirus vector; Handbook Available from: http://ghr.nlm.nih.gov/handbook/illustrations/therapyvectorAccessed June 17, 2015