
Abstract
Background
Human hemoglobin of G-Makassar and hemoglobin E (Hb E) are hemoglobin variants that affect Beta (β) globin. Hb G-Makassar is a very rare variant while Hb E is estimated to affect at least one million people worldwide. Both Hb G-Makassar and Hb E can be inherited in the heterozygous, homozygous or compound heterozygous state. This case series describes the characteristics of four individuals with compound heterozygosity for Hb G-Makassar/Hb E cases in Malaysia. To the best of our knowledge, these are the only four individuals with this genotype reported in the literature.
Case Series
We present four cases of compound heterozygosity for Hb G-Makassar/Hb E identified from October 2014 to January 2021. All the cases were incidental findings whereby the screening Hb analysis showed the presence of peaks in both Hb S and Hb E zones on capillary electrophoresis (CE) and cation-exchange high-performance liquid chromatography (HPLC). Molecular analysis confirmed the findings of compound heterozygous Hb G-Makassar/Hb E. Two cases had a history of anemia secondary to unrelated conditions that resolved with treatment of the underlying cause. The other two cases were asymptomatic individuals who were detected through Malaysia’s National Thalassemia Screening program. On the last follow-up, all the individuals were well, non-transfusion dependent, and had no reported history of chronic anemia, bleeding, hemolysis or thromboembolism complications.
Conclusion
The cases reported here highlight the possibilities for rare compound heterozygous states in multi-ethnicity populations such as Malaysia. Compound heterozygous Hb G-Makassar/Hb E individuals are clinically silent with laboratory values suggesting microcytic and hypochromic red blood cells. Further local epidemiology or population studies with genotyping tests are required for a better understanding of the diversity of its clinical phenotype.
QR Code

Point your SmartPhone at the code above. If you have a QR code reader the video abstract will appear. Or use:
© 2024 Hamzah et al. This work is published by Dove Medical Press Limited, and licensed under Creative Commons Attribution – Non Commercial (unported, v3.0) License. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed.
Introduction
Thalassemia and hemoglobinopathies are the most prevalent genetic disorders and one of the major public health problems in Malaysia. About 4.5% of Malaysian populations are heterozygous carriers for beta (β)-thalassemia.Citation1 Βeta-thalassemia major is the most significant thalassemia with severe anemia requiring life-long blood transfusions for survival.Citation2,Citation3 Among β-thalassemia, Hb E (β26; Glu-Lys) is the second most common abnormal structural variant of hemoglobin in the world after sickle cell hemoglobin (Hb S) and the most common variant in Southeast Asia.Citation4,Citation5 Hb E is prevalent in Malaysia, accounts for 76.0% of the β-thalassemia mutations,Citation6 whereas Hb G-Makassar (β6; Glu-Ala) is very rare. Hb G-Makassar affects the same codon as Hb S (β6; Glu-Val), thus indistinguishable from Hb S by routine Hb analysis methods such as high-performance liquid chromatography (HPLC), capillary electrophoresis (CE), or isoelectric focusing (IEF).Citation7,Citation8
Case Presentation
We describe a total of four individuals with compound heterozygous Hb G-Makassar/Hb E diagnosis from the various states of Malaysia.
Case 1
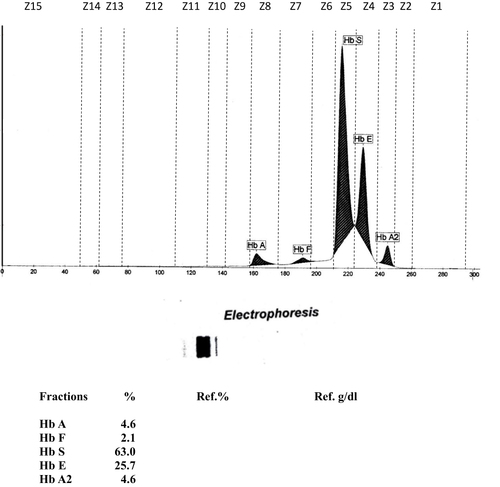
The first case was a 45-year-old male with a history of lower gastrointestinal bleeding secondary to haemorrhoid. This case was reported by Mohamad et al.Citation7 He presented with anemic symptoms and complicated with iron deficiency anemia as a result of frequent bleeding haemorrhoids that required blood transfusions. His initial hematological parameters showed severe anemia with hypochromic microcytic red blood cells (RBCs) with Hb 5.7g/dl, mean corpuscular volume (MCV) 68.9fl and mean corpuscular hemoglobin (MCH) 20.2pg. During hospitalization, an incidental finding of a compound heterozygous Hb S/Hb E was suggested from CE (). However, the genotyping of the β-globin gene had identified a compound heterozygous Hb G-Makassar/Hb E. His anemic symptoms improved after receiving iron therapy and haemorrhoidectomy. The blood count parameter during the Hb analysis is shown in . On follow-up evaluation at 57 years of age, he remained clinically well with no bleeding recurrence since his haemorrhoidectomy with normal hemoglobin concentration of 13.9g/dL and normal serum iron and ferritin level.
Table 1 Demographic, Haematological Parameters, Iron Level and Alpha Mutation Study in All Cases with Compound Heterozygous Hb G-Makassar/Hb E in Malaysia
Figure 1 Hb-electrophoresis chromatogram of Case 1 shows the presence of the Hb variant suggestive of Hb S and Hb E with their percentage.
Case 2
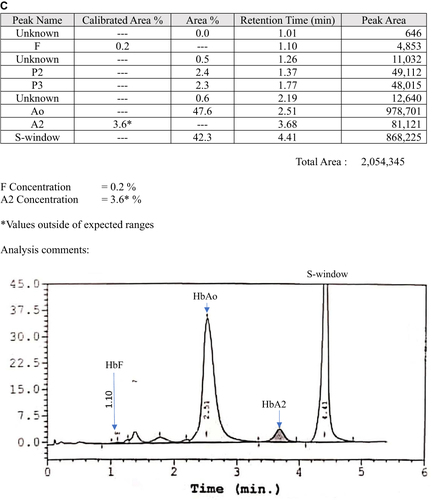
The second case was a 12-month-old boy with underlying congenital hypothyroidism who was found to have a borderline low Hb during admission for fever. He had mild microcytic hypochromic anemia with a normal serum iron level (). The Hb analysis for both CE and HPLC were suggestive of compound heterozygous Hb S/Hb E, nevertheless the β-globin deoxyribonucleic acid (DNA) analysis confirmed compound heterozygous Hb G-Makassar/Hb E. He was euthyroid after treatment with L-thyroxine for three years. At the latest follow-up at the age of 6 years in 2019, he was well with Hb 12.6g/dl, RBC 5.5 × 1012/L, MCV 68.1fl, MCH 22.8pg, MCHC 33.5g/dL and RDW 14.8%. He has normal development for his age and no longer on the follow-up during the phone interview in early 2022. He also has a family history of hemoglobin variants whereby the father and the brother are heterozygous Hb E. Hb analysis using the automated HPLC revealed raised Hb A2 that is suggestive of Hb E ( and ). His mother is heterozygous Hb G-Makassar. Automated HPLC showed a peak at S-window suggestive of Hb S, but DNA analysis confirmed the diagnosis of heterozygous Hb G-Makassar (). Unfortunately, the HPLC chromatogram for Case 2 is not available.
Figure 2 Continued.
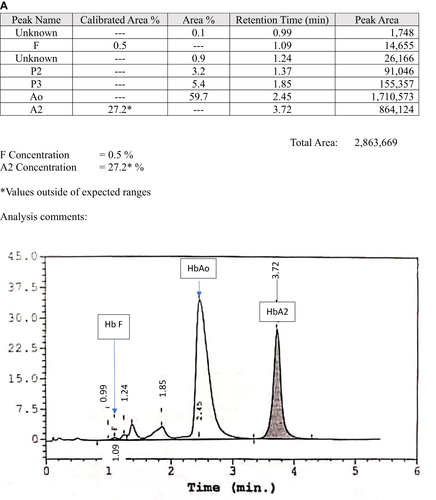
Figure 2 Continued.
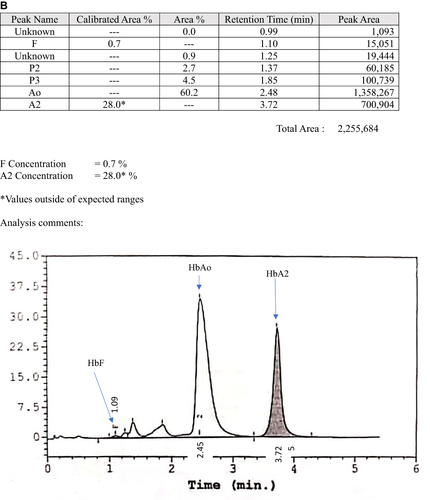
Figure 2 High-performance liquid chromatography (HPLC) analysis of family members of Case 2. (A) Hb E heterozygous father; (B) Hb E heterozygous brother; (C) Hb G-Makassar heterozygous mother.
Case 3 and Case 4
The third and fourth cases were a 16-year-old female and a 16-year-old male, respectively. They were noted to have microcytic hypochromic features although the Hb was within the normal range during Malaysia’s National Thalassemia Screening program. Hb analysis screening for both CE and HPLC were suggestive of compound heterozygous Hb S/Hb E. However, the β-globin DNA analysis identified compound heterozygous Hb G-Makassar/Hb E. Further history in year 2022 at the age of 20 and 19 years old, respectively, revealed they were asymptomatic and denied any symptoms of anemia, history of transfusion-dependence, or features of hemolysis or thrombosis. The third case had normal renal and liver function tests while these tests were not done for the fourth case.
At the time of the study, all the individuals with compound heterozygous Hb G-Makassar/Hb E were alive, asymptomatic, and well. None has any complications such as chronic anemia, hemolysis, transfusion-dependency, thromboembolism, bleeding, severe infections, or hematological malignancy.
Laboratory Investigations
The definitive diagnosis for all the cases was done by direct DNA sequencing analysis of the HBB gene using ABI 3730XL DNA Analyser (Applied Biosystems, Foster City, CA, USA) to detect β-globin variants including Hb G-Makassar and Hb S simultaneously. The laboratory characteristics of compound heterozygous Hb G-Makassar/Hb E individuals are detailed in . DNA analysis for Alpha (α) mutation was performed by multiplex Amplification Refractory Mutation System polymerase chain reaction (ARMS-PCR) and multiplex gap-PCR to detect the common non-deletional and deletional α-gene respectively. The test was performed in view of lower value of presumptive Hb E fraction, low MCV (median 68.4fl, range 61fl - 73.9fl) and MCH (median 23.8pg, range 20.6pg–26.8pg) for all the four individuals but did not reveal any mutations. Among them, only MCV and MCH showed persistently lower than reference ranges. Case 1 had a normal Hb (improved after the treatment given) and a low iron level at screening. He was then asymptomatic with normal blood parameters after undergoing surgical repair for hemorrhoids and correction of iron deficiencies (except MCV persistently < 80fl). All of them showed microcytic hypochromic red cells but absent of sickle cells or features of hemolysis on the peripheral blood smear that would be seen in people with Hb S. We noted that the RBC was normal in Cases 1 and 2 but high in Cases 3 and 4. We postulated that Case 1 and Case 2 had iron deficiency anemia on presentation while Case 3 and Case 4 were normal leading to this discrepancy. Reticulocytes and total bilirubin were not raised in Case 1, 2 and 3 but not tested in Case 4.
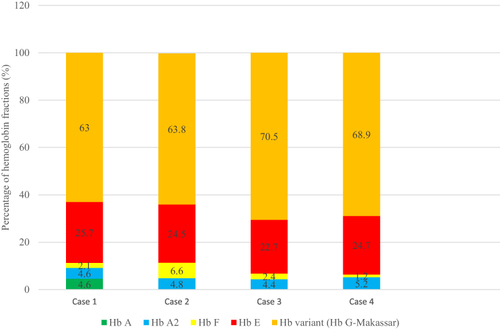
As shown in , Hb G-Makassar level was high within this cohort with a mean of 66.6% ± 3.7. The Hb E level ranged from 22.7% to 25.7% of the total hemoglobin with a mean of 24.4% ± 1.2 and 27.0% ± 1.9 on CE and HPLC, respectively. We assumed the Hb A (4.6%) level in case 1 was from the donor as he received transfusion prior to the screening. There was no concomitant α-thalassemia in these individuals.
Figure 3 Hemoglobin fractions by capillary electrophoresis of four individuals with compound heterozygous Hb G-Makassar/Hb E. We assumed the Hb A (4.6%) level in case 1 was from the donor as he received a transfusion prior to the screening.
Discussion
Hb G-Makassar is a nonpathological β-chain variant characterized by a single nucleotide substitution GAG-> GCG at the β6 or A3 position that changes normal glutamyl residue to alanyl residue (β6 Glu→Ala). This rare mutation was first identified in Makassar, Sulawesi (Celebes), the Republic of Indonesia in 1969.Citation9 Hb G-Makassar heterozygotes and homozygotes are asymptomatic with normal complete blood count parameters though its structural change occurs at the same position as that in Hb S.Citation10 Furthermore, Hb G-Makassar appears to have the properties of normal Hb A and does not polymerize or lead to hemolysis like sickle cells.Citation11,Citation12 In contrast, the Hb G-Makassar/β0-thalassemia compound heterozygote has hematologic features of thalassemia trait.Citation8
Hb E gene is a mutant form of the β-globin gene that encodes lysine instead of glutamate at position 26. This β-E chain is inefficiently produced because of a novel cryptic messenger RNA splicer site, leading to thalassemic RBC indices such as microcytosis.Citation13,Citation14 Individuals with homozygous Hb E (Hb EE) and Hb E trait are clinically normal but compound heterozygous Hb E/β+ or Hb E/β0 thalassemia may have clinical manifestations of thalassemia, including compound heterozygosity for Hb E and Hb S (Hb SE).Citation15–18 Hb SE may result in sickle cell crises such as vaso-occlusive pain crises and hemolytic anemia, particularly during exposure to stress conditions or deoxygenated states such as infection or hypoxemia, unlike compound heterozygous Hb G-Makassar/Hb E, which is essentially asymptomatic. It is important to distinguish Hb E disorders diagnostically as it has different clinical courses amongst different genotypes. For example, Hb E beta-thalassemia (Hb E/β) has a wide phenotype that ranges from mild anemia to severe transfusion-dependent thalassemia major.Citation19 Compound heterozygous Hb G-Makassar/Hb E in our cohort can be considered mild phenotype of β-thalassemia as they are clinically normal though having some thalassemia trait red cell indices.
In Malaysia, Hb E/β-thalassemia (34.4%) is the most common form of β-thalassemia and majority in Malay ethnicity, followed by β-thalassemia major (33.5%), hemoglobin H (Hb H) disease (18.3%), β-thalassemia intermedia (9.4%), and ‘others’ (4.5%). The ‘others’ group includes other forms of Hb H disease, Hb Lepore Hollandia, α-thalassemia syndrome, δβ-thalassemia, and other thalassemia disorders requiring regular blood transfusions.Citation20 However, the prevalence and incidence of Hb G-Makassar including compound heterozygous Hb G-Makassar/Hb E remain less elucidative. The incidence of both Hb E and Hb G-Makassar could be underrepresented as they are usually asymptomatic and red cell indices are commonly unremarkable on a routine blood counts check-up, hence are not likely to be sent for screening. Nevertheless, Hb E can be easily detected on Hb electrophoresis screening but the incidence of Hb G-Makassar may be underestimated as DNA molecular analysis is required for diagnostic confirmation. Therefore, in asymptomatic individuals with borderline or lowish MCV/MCH levels who are suspected to have Hb E and Hb S peaks on HPLC or CE, we suggest they also undergo genotyping.
Some genetic modifiers and prognostic indicators affect the severity of phenotype, including type of β-chain mutation, Hb F levels, concomitance of α-thalassemia, age of presentation and splenomegaly. Thromboembolism risk is high in post-splenectomised thalassemia patients due to a hypercoagulable state.Citation21 Morbidity from iron overload in non-transfused patients is also commonly seen, secondary to increased gastrointestinal iron absorption.Citation22 Hb E/β and Hb SE patients require regular monitoring because of risk of cardiopulmonary disease such as pulmonary hypertension and cardiac failure secondary to iron overload, chronic thromboembolism, and hemolysis-induced nitric oxide deficiency.Citation14,Citation23–25 The laboratory characteristics and the phenotypic variability in compound heterozygous Hb G-Makassar/Hb E individuals are not widely reported. As opposed to Hb Eβ and HbE/S, all our cases of compound heterozygous Hb G-Makassar/Hb E were well with no clinical symptoms and did not show any complications during individual follow-up. None were on any treatment or needs regular follow-up. Our cases clearly show that compound heterozygous Hb G-Makassar/Hb E is a mild phenotype of hemoglobinopathy and asymptomatic.
For the laboratory tests, our compound heterozygous Hb G-Makassar/Hb E individuals showed unremarkable haematological parameters except for low MCV and MCH. The low MCV < 74fl, MCH < 24pg and lower Hb E < 25–26% are likely suggestive of Hb E concomitant α-thalassemia traitCitation26,Citation27 and Hb E level <24% was reported superior to MCV and MCH for differentiating the heterozygous Hb E with or without α0-thalassemia trait.Citation28 Interestingly, none of our cases had α mutations. Hb E percentage also can be reduced by iron deficiency. However, we could not conclude coexisting iron deficiency as not all individuals did iron studies. The high level of Hb G-Makassar (>60%) could explain the mild phenotype in our cohort as there is preferential binding of α-globin chain subunit to Hb G-Makassar than Hb E (range 22.7% to 25.7%). The Hb E level on CE was lower than HPLC and is consistent with the study by Hafiza et al. This is due to the Hb E and HbA2 coeluted at the same retention time on HPLC meanwhile CE measured the actual level of Hb E in the sample.Citation29 On alkaline Hb electrophoresis, Hb E migrates with C, O Arab and A2 whilst in acid pH electrophoresis testing, it migrates with Hb A2.
Hb G-Makassar has been reported as a promising new treatment target that may assist in gene editing therapy in sickle cell disease patients based on installing the Hb G-Makassar variant to replace the pathogenic Hb S allele.Citation30–32 The potential for this approach to be used to treat sickle cell disease (SCD) in humans is an area of active investigation. Recently, non-clinical studies in homozygous Hb S cells showed that gene editing of the pathogenic Hb S allele into Hb G-Makassar with an adenine base editor led to normal hemoglobin function in vitro and rescued mice with sickle cell disease in vivo.
Therefore we suggest a local epidemiology or population studies with genotyping tests to elucidate the status of Hb G-Makassar in the future for a better understanding of the diversity of its clinical phenotype.
Conclusion
The cases reported here highlight the possibilities for rare compound heterozygous states in multi-ethnicity populations such as Malaysia. Individuals with compound heterozygosity for Hb G-Makassar and Hb E had laboratory values demonstrating microcytic and hypochromic RBCs but were clinically asymptomatic without evidence of chronic anemia, hemolysis, bleeding or thrombosis. These findings may provide a valuable reference for prenatal diagnosis and genetic counseling as well as data to support sickle cell disease therapy based on installing the Hb G-Makassar variant to replace the pathogenic Hb S allele.
Abbreviations
Hb, Hemoglobin; CE, Capillary electrophoresis; HPLC, Cation-exchange high-performance liquid chromatography; IEF, Isoelectric focusing; RBCs, Red blood cells; MCV, Mean corpuscular volume; MCH, Mean corpuscular hemoglobin; MCHC, Mean cell hemoglobin concentration; DNA, Deoxyribonucleic acid; RDW, Red cell distribution width.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Declaration of Parental Consent
Ethical approval to report this case series was obtained from Medical Research and Ethics Committee, (NMRR-21-1408-60675, IIR), National Institutes of Health, Ministry of Health Malaysia, Malaysia. The authors certify that they have obtained all appropriate patient consent forms. In the form the parental(s) has/have given his/her/their consent for his/her/their child images and other clinical information to be reported in the journal. The parents understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed. The case 1 patient in this study has given written consent to participate as well as consent to publish his data.
Consent for Publication
This study was conducted in accordance with the fundamental principles of the Declaration of Helsinki.
Disclosure
Guo Chen is an employee and stockholder of Beam Therapeutics Inc. The authors report no other conflicts of interest in this work.
Acknowledgments
This study was initiated and supported by Beam Therapeutics Inc. We thank all individuals and staff for their support and for participating in this study including Dr. Jameela Sathar and Dr. Tan Sen Mui from Department of Haematology, Ampang Hospital, Dr. Zaihawa Hamad from Department of Pathology, Putrajaya Hospital, Putrajaya, Dr. Norhafizatul Aida Amir Khalid from Medical Department, Hospital Sultanah Nur Zahirah, Terengganu, Malaysia and Mohammad Azannee Saad from International Islamic University Malaysia.
References
- George E. Thalassaemia Carrier Diagnosis in Malaysia. Thalassaemia Diagnostic Service; 1998.
- Orkin SH, Kazazian HH. The mutation and polymorphism of the human β-globin gene and its surrounding DNA. Ann Rev Gene. 1984;18(1):131–171. doi:10.1146/annurev.ge.18.120184.001023
- Mary Anne Tan JA, Chin PS, Wong YC, Tan KL, Chan LL, George E. Characterisation and confirmation of rare beta‐thalassaemia mutations in the Malay, Chinese and Indian ethnic groups in Malaysia. Pathology. 2006;38(5):437–441. doi:10.1080/00313020600922538
- Fucharoen S, Winichagoon P. Hemoglobinopathies in Southeast Asia: molecular biology and clinical medicine. Hemoglobin. 1997;21(4):299–319. doi:10.3109/03630269709000664
- Weatherall D. The inherited disorders of haemoglobin: an increasingly neglected global health burden. Indian J Med Res. 2011;134(4):493.
- Alwi ZB, Syed-Hassan S-NR-K. Thalassemia in Malaysia. Hemoglobin. 2022;46(1):45–52. doi:10.1080/03630269.2022.2057326
- Mohamad AS, Hamzah R, Selvaratnam V, Yegapan S, Sathar J. Human hemoglobin G-Makassar variant masquerading as sickle cell anemia. Hematol Rep. 2018;10(3):7210. doi:10.4081/hr.2018.7210
- Viprakasit V, Wiriyasateinkul A, Sattayasevana B, Miles KL, Laosombat V. Hb G-makassar [β 6 (A3) Glu→ Ala; codon 6 (GAG→ GCG)]: molecular characterization, clinical, and hematological effects. Hemoglobin. 2002;26(3):245–253. doi:10.1081/HEM-120015028
- Blackwell RQ, Oemijati S, Pribadi W, Weng M-I, Liu C-S. Hemoglobin G Makassar: β6 Glu→ Ala. Biochimica Et Biophysica Acta (BBA)-Protein Structure. 1970;214(3):396–401. doi:10.1016/0005-2795(70)90297-7
- Sangkitporn S, Mitrakul C, Rerkamnuaychoke B, Sutivigit Y, Sangkitporn S. Hb G Makassar (beta 6: glu→ ala) in a Thai family. J Med Assoc Thai. 2002;85(5):577–582.
- Adachi K, Kim J, Konitzer P, Asakura T, Saviola B, Surrey S. Effects of β6 amino acid hydrophobicity on stability and solubility of hemoglobin tetramers. FEBS Lett. 1993;315(1):47–50. doi:10.1016/0014-5793(93)81130-R
- Pagnier J, Bihoreau M, Baudin V, Edelstein S, Poyart C. Polymerization and solubility of recombinant hemoglobins alpha 2 beta 2 6 Glu--> Ala (Hb Makassar) and alpha 2 beta 2 6 Glu--> Ala, 23 Val--> Ile. Comptes Rendus de L’academie des sciences Serie III, Sciences de la vie. 1993;316(4):431–436.
- Benz EJ, Berman BW, Tonkonow BL, et al. Molecular analysis of the beta-thalassemia phenotype associated with inheritance of hemoglobin E (alpha 2 beta2(26)Glu leads to Lys). J Clin Invest. 1981;68(1):118–126. doi:10.1172/JCI110226
- Fucharoen S, Winichagoon P. Clinical and hematologic aspects of hemoglobin E β-thalassemia. Current Opinion Hematol. 2000;7(2):106–112. doi:10.1097/00062752-200003000-00006
- Fucharoen S, Winichagoon P. Hemoglobinopathies in Southeast Asia. Hemoglobin. 1987;11(1):65–88. doi:10.3109/03630268709036587
- Tyagi S, Pati H, Choudhry V, Saxena R. Clinico-haematological profile of HbE syndrome in adults and children. Hematology. 2004;9(1):57–60. doi:10.1080/10245330310001638983
- Mishra P, Pati H, Chatterjee T, et al. Hb SE disease: a clinico-hematological profile. Ann Hematol. 2005;84(10):667–670. doi:10.1007/s00277-005-1044-2
- Hanafi SB, Abdullah WZ, Adnan RA, et al. Genotype-phenotype association of HbE/β-thalassemia disease and the role of genetic modifiers. Malays J Paediatr Child Health. 2016;22:1–16.
- Olivieri NF, Pakbaz Z, Vichinsky E. Hb E/beta-thalassaemia: a common & clinically diverse disorder. Indian J Med Res. 2011;134(4):522.
- Ibrahim HM, Muda Z, Othman IS, et al. Observational study on the current status of thalassaemia in Malaysia: a report from the Malaysian Thalassaemia Registry. BMJ open. 2020;10(6):e037974. doi:10.1136/bmjopen-2020-037974
- Vichinsky E. Hemoglobin e syndromes. Hematology Am Soc Hematol Educ Program. 2007;2007(1):79–83. doi:10.1182/asheducation-2007.1.79
- Pakbaz Z, Fischer R, Fung E, Nielsen P, Harmatz P, Vichinsky E. Serum ferritin underestimates liver iron concentration in transfusion independent thalassemia patients as compared to regularly transfused thalassemia and sickle cell patients. Pediatr Blood Cancer. 2007;49(3):329–332. doi:10.1002/pbc.21275
- Morris CR, Kuypers FA, Kato GJ, et al. Hemolysis‐associated pulmonary hypertension in thalassemia. Ann NY Acad Sci. 2005;1054(1):481–485. doi:10.1196/annals.1345.058
- Eldor A, Rachmilewitz EA. The hypercoagulable state in thalassemia. Blood. 2002;99(1):36–43. doi:10.1182/blood.V99.1.36
- Atichartakarn V, Chuncharunee S, Chandanamattha P, Likittanasombat K, Aryurachai K. Correction of hypercoagulability and amelioration of pulmonary arterial hypertension by chronic blood transfusion in an asplenic hemoglobin E/β-thalassemia patient. Blood. 2004;103(7):2844–2846. doi:10.1182/blood-2003-09-3094
- Sanchaisuriya K, Chirakul S, Srivorakun H, et al. Effective screening for double heterozygosity of Hb E/α0-thalassemia. Ann Hematol. 2008;87(11):911–914. doi:10.1007/s00277-008-0520-x
- Charoenkwan P, Wanapirak C, Thanarattanakorn P, et al. Hemoglobin E levels in double heterozygotes of hemoglobin E and SEA-type alpha-thalassemia. Southeast Asian J Trop Med Public Health. 2005;36(2):467–470.
- Pornprasert S, Tookjai M, Punyamung M, Pongpunyayuen P. HbE Level and Red Cell Parameters in Heterozygous HbE With and Without α0-Thalassemia Trait. Indian J Hematol Blood Transfusion. 2018;34(4):662–665. doi:10.1007/s12288-018-0947-8
- Alauddin H, Khirotdin RA, Ithnin Z, et al. HbA2 levels in normal,[Beta]-thalassaemia and haemoglobin E carriers by capillary electrophoresis. Malays J Pathol. 2012;34(2):161.
- Chu SH, Ortega M, Feliciano P, et al. Conversion of HbS to Hb G-makassar by adenine base editing is compatible with normal hemoglobin function. Blood. 2021;138(Supplement 1):951. doi:10.1182/blood-2021-150922
- Chu SH, Packer MS, Olins JR, et al. A novel base editing approach to directly edit the causative mutation in sickle cell disease. Mol Ther. 2020;28:808.
- Newby GA, Yen JS, Woodard KJ, et al. Base editing of haematopoietic stem cells rescues sickle cell disease in mice. Nature. 2021;595(7866):295–302. doi:10.1038/s41586-021-03609-w