
Abstract
Purpose
To analyze the composition of abnormal hemoglobin and the relationship between genotype and phenotype by screening abnormal hemoglobin in a subpopulation of Guizhou, China.
Patients and Methods
Routine blood evaluation, capillary electrophoresis of hemoglobin, and mutation of α - and β - thalassemia genes were evaluated in 19,976 individuals for thalassemia screening in Guizhou. Sanger sequencing of HBA1, HBA2 and HBB genes was performed in samples with abnormal bands or unexplained increases of normal bands. The types of abnormal hemoglobin were obtained by sequence analysis.
Results
Abnormal hemoglobin was detected in 84 individuals (detection rate, 0.42%). Ten types each of α and β globin chain variants were detected, including most commonly Hb E, Hb New York and Hb Port Phillip. In this study, the abnormal Hb Mizuho was identified for the first time in a Chinese population, and a novel abnormal hemoglobin Hb Guiyang (HBA2: c.151C > A) was detected for the first time. Except for Hb Mizuho, other abnormal hemoglobin heterozygotes without thalassemia or iron deficiency had no significant hematological changes.
Conclusion
This study enriched the molecular epidemiological data of abnormal hemoglobin in Guizhou, China and provided reference data for genetic counseling and prenatal diagnosis of abnormal hemoglobin.
Introduction
Hemoglobinopathy is a type of autosomal monogenic genetic disease with a globin gene mutation that results in insufficient hemoglobin synthesis or structural abnormalities, which is widely distributed worldwide, especially in the Mediterranean, Africa and Southeast Asia.Citation1 Approximately 7% of the world’s population are hemoglobinopathy carriers, and 330,000 newborns are born each year with hemoglobinopathy (91%, sickle cell disease), which causes 3.4% of deaths in children under the age of 5.Citation2 Clearly, this disorder poses a major worldwide public health problem.
Hemoglobinopathies are divided into two major categories, one is abnormal hemoglobin caused by mutations in the globin gene resulting in abnormal structure of the globin chain, also known as hemoglobin variant, an example of which is sickle cell disease. The other is inhibition of globin peptide chain synthesis caused by defects or mutations in the globin gene, resulting in chronic hemolytic anemia called thalassemia.Citation3 According to the human hemoglobin variants and thalassemia mutation database HbVar (https://globin.bx.psu.edu/hbvar/), 1429 hemoglobin variants have been identified, and this number is still rising. Epidemiological data from various localities have shown that there are ethnic and geographical differences in the incidence of hemoglobinopathy and the distribution of mutation types.Citation4 In most hemoglobinopathy-endemic populations, α- and β-thalassemia coexist with various abnormal hemoglobins.Citation5 Abnormal hemoglobin causes symptoms in individuals ranging from almost asymptomatic to severe hemolytic anemia and the hemoglobin molecule is characterized by structural abnormalities due to changes in the conformation of the globin peptide chain. Although the mutation types include base deletion and insertion, frameshift mutation, stop code mutation, fusion genes resulting from exchange between different genes, the vast majority are single amino acid substitutions caused by point mutations. Although most abnormal hemoglobins have limited clinical significance, a small number of homozygotes or if combined with thalassemia will present with significant anemia, such as Hb S/S and Hb E/β0.Citation6,Citation7
The study of hemoglobinopathy in China was initiated in the 1960s, and in the 1980s, Chinese scientists conducted a general survey of hemoglobinopathy in nearly one million people covering 28 provinces nationwide. That survey showed that the average carrier rate of abnormal hemoglobin in Chinese population was 0.33%, and the distribution showed a significant difference between the south (0.37%) and the north (0.29%).Citation8 Although genotype identification was not performed then due to technical reasons, with the rapid development of sequencing technology, abnormal hemoglobin genotypes have been continuously identified throughout China. Because the carrier rate of abnormal hemoglobin is much lower than thalassemia, and often asymptomatic, only a few abnormal hemoglobins have been identified when the indication was thalassemia screening and gene sequencing prompted by thalassemia-like clinical manifestations. Southwest China is known as a high incidence area of thalassemia, with many reports of thalassemia in Guizhou, but there have been no reports of a large-scale investigation of abnormal hemoglobin phenotype and genotype. The aim of this study, therefore, was to investigate the phenotype and genotype of abnormal hemoglobin in a large cohort of people in Guizhou, China.
Materials and Methods
Subjects
The study cohort comprised 19,976 individuals from Guizhou Province, enrolled from October 2019 to January 2023, who underwent thalassemia screening at Guizhou Provincial People’s Hospital. Three tubes of EDTA-anticoagulated venous blood were collected from each individual for routine blood evaluation, hemoglobin electrophoresis, and detection of common α and β thalassemia gene mutation types.
Hematology Test
Routine blood examination was performed using an automatic hematology analyzer (Sysmex, XN-9000, Kobe Japan), and the hemoglobin concentration (Hb), erythrocyte mean corpuscular volume (MCV) and mean corpuscular hemoglobin (MCH) were recorded. Hemoglobin electrophoresis was performed using an automatic hemoglobin capillary electrophoresis instrument (Sebia, Capillarys2, Paris, France) to assess the concentrations of Hb A, Hb A2, Hb F and any abnormal Hbs.
Thalassemia Gene Detection
Genomic DNA was extracted from 2 mL of EDTA anticoagulated blood with TIANamp Genomic DNA Kit (Tiangen Biotech,TIANGEN, Beijing, China) according to the manufacturer’s recommendations. α and β thalassemia gene detection kits and matched automatic nucleic acid hybridization instruments (Hybriobio Limited, HBHM-3000S, Guangzhou, China) were used to detect six common α-thalassemia mutations (--SEA, -α3.7, -α4.2, Hb CS CD142 TAA > CAA, Hb QS CD125 CTG > CCG, Hb WS CD122 CAC > CAG) and 17 β-thalassemia mutations (CD41-42 -TCTT, CD43 G > T, IVS-II-654 C > T, CD17 A > T, CD14-15 + G, −28 A > G, −29 A > G, CD71-72 + A, βE G > A, IVS-I-1 G > A/T, CD27-28 + C, IVS-I-5 G > C, CAP A > C or -AAAC, Int T > G, CD31-C, −30 T > C and −32 C > A).
DNA Sequencing
Sanger sequencing of HBA1, HBA2 and HBB genes was performed in those with abnormal bands or increased normal band content on electrophoresis. From the Genbank database (www.ncbi.nlm.nih.gov), the reference sequences of HBA1, HBA2, and HBB were obtained, and primers were designed using Primer 5.0 software and synthesized by BiOligo Biotechnology (BiOligo Biotech, Shanghai, China) to amplify the HBA1, HBA2, and HBB genes, respectively. Primer sequences are shown in . The amplified products were sequenced with a sequencer (ABI, 3730XL, MA, USA), and then the sequencing results were compared with the reference sequences to identify the mutations leading to abnormal hemoglobin.
Table 1 Primers Used for Sanger Sequencing of HBA1, HBA2 and HBB Gene
Pathogenicity Analysis of Emerging Abnormal Hemoglobin
Using Clustal omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) for conservative mutation amino acid analysis, and using Phyre2 (http://www.sbg.bio.ic.ac.uk) and SWISS-PDB Viewer4.10 for protein three dimensional structure prediction and analysis, the sample was analyzed for pathogenicity according to ACMG (American College of Medical Genetics and Genomics) guidelines.
Results
Of 19,976 individuals screened for thalassemia, 84 cases (detection rate, 0.42%) of abnormal hemoglobin were detected, all of which were heterozygous. There were 15 cases (18%) of α-globin variants which included 10 types: Hb Port Phillip, Hb I, Hb Orbassano, Hb Q-Thailand, Hb J-Toronto, Hb J-Norfolk, Hb G-Waimanalo, Hb Beijing, Hb Hekinan II and Hb Guiyang; Hb Port Phillip was the most common. The positions of HBA gene mutations, amino acid changes, abnormal hemoglobin electrophoresis bands, and contents of α-globin variants are shown in .Hematological characteristics of hemoglobin with abnormal α globin chain are shown in . Sixty-nine cases (82%) of β-globin variation were detected, which included 10 types: Hb E, Hb New York, Hb J Bangkok, Hb D-Punjab, Hb Lome, Hb G-Taipei, Hb G-San José, Hb Hope and Hb Mizuho; Hb E (50/84) was the most common, followed by Hb New York (10/84). The positions of HBB gene mutations, amino acid changes, abnormal hemoglobin electrophoresis bands and contents of β-globin variants are shown in .Hematological characteristics of hemoglobin with abnormal β globin chain are shown in .
Table 2 α Variation Types, Electrophoresis Bands and Average Content of Globin Chain
Table 3 Hematological Characteristics of Hemoglobin with Abnormal α Globin Chain
Table 4 β Variation Types, Electrophoresis Bands and Average Content of Globin Chain
Table 5 Hematological Characteristics of Hemoglobin with Abnormal β Globin Chain
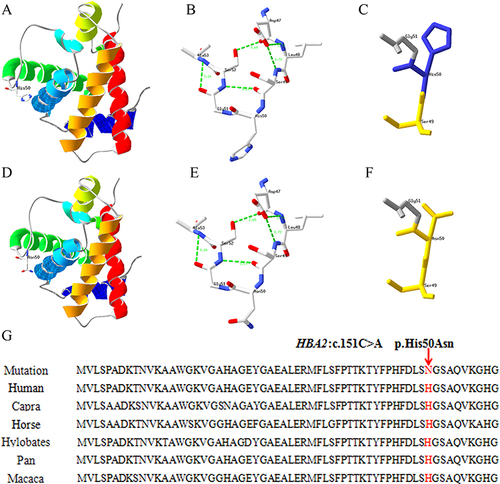
In the present study, a novel abnormal hemoglobin (HBA2: c.151C > A) was detected, named Hb Guiyang because it was first identified in Guiyang city. The individual with Hb Guiyang was a 29-year-old woman who had red cell indices of RBC 4.45×1012/L, Hb 136 g/L, MCV 90.6 fL, MCH 30.6 pg; hemoglobin electrophoresis results of HbA 74.3%, HbA2 2.1%, and abnormal hemoglobin 23.6% in Z12 region (). Her thalassemia screening genotypes for α- and β-thalassemia were αα/αα and βN/βN, respectively, and Sanger sequencing results showed that she was a heterozygous carrier of HBA2: c.151C > A (). The conserved analysis of mutant amino acids in six species was highly conserved, and the three-dimensional structure prediction of the protein showed no change in hydrogen bonds and only charge changes (). The pathogenicity of HBA2: c.151C > A was a variant of uncertain significance (VUS) as evidenced by PM2 according to the ACMG guidelines. In addition, one case of Hb Mizuho (HBB: c.206T >C) was detected in a 2-year-old girl with severe hemolytic anemia, the first time Hb Mizuho was found in a Chinese population.
Figure 1 Hemoglobin capillary electrophoresis of the patient with Hb Guiyang.
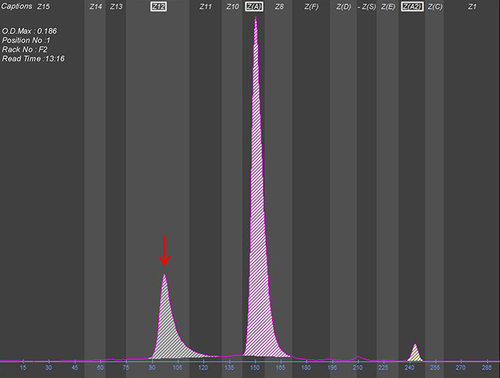
Figure 2 Sanger sequencing peak of HBA2 gene in patients with Hb Guiyang.
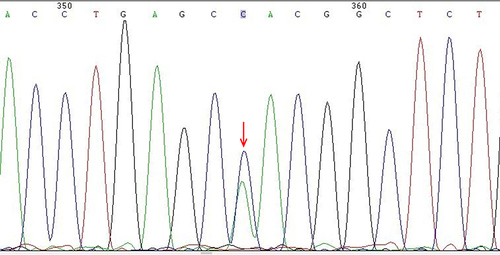
Figure 3 Protein three-dimensional structure prediction and conservation analysis of the Hb Guiyang. (A, B and C) are the three dimensional structure prediction and the partial enlargement of wild-type protein. (D, E and F) are the mutant protein. (G): the positions p.His50 in the HBA2 protein are highly conserved among six species.
Among the 15 cases of abnormal α-chain hemoglobin, 12 were heterozygotes only, none of whom had clinical manifestations, their Hb content was above 110 g/L, and both their MCV and MCH were in the normal range. Two heterozygote cases of abnormal α-chain hemoglobins with silent α-thalassemia had no anemia phenotype, but their MCV and MCH were below the normal range.The study identified a novel mutation for α-thalassaemia and Hb Portland that is not common in Southwest China. One heterozygote case of abnormal α-chain hemoglobin with iron deficiency anemia had significant anemia manifestations (Hb: 62 g/L), and MCV and MCH were also below the normal range.
Among the 69 cases of abnormal β-chain hemoglobin, 55were heterozygotes only, including 41 Hb E heterozygotes, and their mean hemoglobin content was normal (121.5 ± 16 g/L), but their MCV and MCH were below the normal range. One heterozygote case of Hb Mizuho had significant manifestations of anemia (Hb: 67 g/L), with both MCV and MCH in the normal range. The other 13 heterozygotes of abnormal β-chain hemoglobin had no clinical phenotype, and their hemoglobin content, MCV and MCH were within the normal range. Seven heterozygote cases of abnormal β-chain hemoglobin with iron deficiency anemia showed moderate to severe anemia, and their MCV and MCH were also below the normal range. There were three heterozygote cases of abnormal β-chain hemoglobin with α-thalassemia minor, of which two cases had no anemia, one case had mild anemia, and the MCV and MCH of three cases were below the normal range. Two Hb E heterozygotes with silent α-thalassemia had no clinical phenotype, and both their MCV and MCH were within the cut-off range. Two Hb E heterozygotes with β-thalassemia minor showed moderate to severe anemia, and their MCV and MCH were below the normal range.
Discussion
In this study, 84 cases of abnormal hemoglobin were detected in 19,976 Guizhou patients with thalassemia screening, including 15 cases of α globin chain and 69 cases of β globin chain, with a total detection rate of 0.42%, higher than the carrier rate of 0.2% in Guizhou in the 1980s.Citation8 It was lower than 0.78% in Yunnan,Citation9 0.59% in Guangxi,Citation10 0.57% in Chongqing,Citation11 0.49% in Hunan,Citation12 and 0.44% (excluding Hb E) in Guangxi, Yunnan-Guizhou junction,Citation13 which may be related to regional and population differences.
According to reports in the 1980s, there were differences in the type of abnormal hemoglobin between the South and North of China; the most common abnormal hemoglobins in the South were Hb E, Hb New York, Hb G Chinese, Hb Q Thailand, and Hb J Bangkok; the most common abnormal hemoglobin in the North was Hb D Punjab.Citation8 In recent years, a large screening sample (311,042 people) in Southern China found the detection rate of abnormal hemoglobin was 0.35% (117 people), and the most common abnormal hemoglobins were: Hb E, Hb New York, Hb J Bangkok, and Hb Q Thailand.Citation14 In the present study, 20 types of abnormal hemoglobin were detected, including 10 kinds each of α and β globin chains, and the three most common abnormal hemoglobins were Hb E (50/84), Hb New York (10/84), and Hb Port Phillip (4/84). Hb E is undoubtedly the most common abnormal hemoglobin in Southern China, followed by Hb New York, consistent with previous reports. Surprisingly, however, four cases of Hb Port Phillip were found in this study, and only a few cases have been reported in the literature worldwide,Citation14–18 indicating that Hb Port Phillip is surprisingly not rare in Guizhou. Additionally, abnormal hemoglobin Hb Mizuho, Hb Orbassano, Hb J-Toronto, and Hb J-Norfolk were also reported for the first time in the Chinese population, and abnormal hemoglobin Hb Beijing and Hb G-San José were reported for the first time in the Guizhou population of China.
By querying the HbVar database, this study found an abnormal hemoglobin (HBA2: c.151C > A) that had not been previously reported worldwide; it was caused by the substitution of histidine by asparagine at codon 50 CAC > AAC of the α2 globin gene. According to international nomenclature, this abnormal hemoglobin was named Hb Guiyang. In this case, the newly identified abnormal hemoglobin carriers had a normal hematological phenotype, and 23.6% of the abnormal peak profiles appeared in the Z12 region only in hemoglobin capillary electrophoresis. Although the conservation analysis of amino acids at the mutation point was highly conserved, protein structure prediction showed that only the charge of amino acids at the mutation point was changed, and amino acids before and after mutation were polar amino acids, consult NCBI (https://www.ncbi.nlm.nih.gov). This mutation site is known not to be located in the position where it binds to the heme moiety, and not at the tetramer contact surface; it is speculated that it does not affect protein structural stability. Combined with the pathogenicity analysis of ACMG, together with the abnormal hemoglobins Hb South Yorkshire (HBA2: c.151C > T) and Hb J-Sardegna (HBA2: c.151C > G) being caused by different base substitutions at the same locus but with no clinical manifestations, in the present case, it was comprehensively speculated that this mutation would not cause abnormal hemoglobin function.
Most abnormal hemoglobins have only mild clinical manifestations or none at all because their mutated amino acids are located outside the hemoglobin molecule. Nevertheless, a few can cause moderate to severe anemia, especially referring to certain abnormal hemoglobin homozygotes, combined thalassemia, or other abnormal hemoglobins. Concordant with this study, Hb E heterozygotes alone have been shown to cause mild microcytic hypochromia with or without anemia, and there were two cases of significant anemia when Hb E was combined with β0, which was due to the fact that Hb E/β0 had little β-chain synthesis, there was little HbA production, and hemoglobin naturally decreased.Citation19,Citation20 One case of Hb Hope complex--SEA/αα showed mild microcytic hypochromic anemia, consistent with Pornprasert’s report.Citation21 Although Hb Port Phillip has been documented on HbVar to cause hemoglobin instability due to loss of the heme interface, Hb Port Phillip heterozygotes alone in this study did not have significant manifestations of anemia and were only significantly anemic when combined with --SEA/αα, consistent with the findings of Du et al.Citation17 Because Hb Port Phillip is rarely reported in the literature, the study of phenotype needs to be further accumulated. From the literature and our previous report,Citation22 Hb Mizuho caused severe hemolytic anemia because the mutated amino acid affected the binding of distal histidine β63 (E7) to the heme moiety. Relative to clinical management of unexplained anemia, DNA sequencing of the globin gene should be performed to identify the cause even if routine hemoglobin component tests reveal no abnormalities.
Conclusion
In summary, this study confirmed that there were a wide variety of abnormal hemoglobin species in Guizhou, China. This study enriched the molecular epidemiological data of abnormal hemoglobinopathy in Guizhou, and also provided reference data for genetic counseling and prenatal diagnosis in Guizhou.
Ethics and Consent Statements
This study was approved by the Medical Ethics Committee at Guizhou Provincial People’s Hospital (approval number 2022-05). We obtained informed consent forms from all subjects or guardians. We confirm that our study complies with the Declaration of Helsinki.
Disclosure
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Acknowledgments
The authors appreciate all individuals participated in this study and appreciate the EditSprings for the expert linguistic services provided.
Additional information
Funding
References
- Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ. 2001;79(8):704–712.
- Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86(6):480–487. doi:10.2471/BLT.06.036673
- Shang X, Xu X. Update in the genetics of thalassemia: what clinicians need to know. Best Pract Res Clin Obstet Gynaecol. 2017;39:3–15. doi:10.1016/j.bpobgyn.2016.10.012
- Kohne E. Hemoglobinopathies: clinical manifestations, diagnosis, and treatment. Dtsch Arztebl Int. 2011;108(31–32):532–540. doi:10.3238/arztebl.2011.0532
- Harteveld C, LAchour A, Arkesteijn SJG, et al. The hemoglobinopathies, molecular disease mechanisms and diagnostics. Int J Lab Hematol. 2022;44(Suppl 1):28–36. doi:10.1111/ijlh.13885
- Lage J, Monteiro B, Costa A, Mendes IF, Ferreira T, Loureiro HC. Acute complications of sickle cell disease in children under 5 years at a level II hospital. Glob Pediatr Health. 2022;9:2333794X221141356. doi:10.1177/2333794X221141356
- Traivaree C, Monsereenusorn C, Rujkijyanont P, Prasertsin W, Boonyawat B. Genotype-phenotype correlation among beta-thalassemia and beta-thalassemia/HbE disease in Thai children: predictable clinical spectrum using genotypic analysis. J Blood Med. 2018;9:35–41. doi:10.2147/JBM.S159295
- Zeng YT, Huang SZ. Disorders of haemoglobin in China. J Med Genet. 1987;24(10):578–583. doi:10.1136/jmg.24.10.578
- Zhang J, Li P, Yang Y, et al. Molecular epidemiology, pathogenicity, and structural analysis of haemoglobin variants in the Yunnan province population of Southwestern China. Sci Rep. 2019;9(1):8264. doi:10.1038/s41598-019-44793-0
- Xiong F, Sun M, Zhang X, et al. Molecular epidemiological survey of haemoglobinopathies in the Guangxi zhuang autonomous region of southern China. Clin Genet. 2010;78(2):139–148. doi:10.1111/j.1399-0004.2010.01430.x
- Li CL, Yang M, Li QH. Analysis of 34 800 cases of abnormal hemoglobinopathy in couples of child-bearing age in Chongqing area. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2020; 28(4):1316–1320.doi:10.19746/j.cnki.issn.1009-2137.2020.04.040.
- Xi H, Liu Q, Xie DH, et al. Epidemiological survey of hemoglobinopathies based on next-generation sequencing platform in Hunan province, China. Biomed Environ Sci. 2023;36(2):127–134. doi:10.3967/bes2023.016.
- Xu G, Wang C, Wang J, et al. Prevalence and molecular characterization of common thalassemia among people of reproductive age in the border area of Guangxi-Yunnan-Guizhou province in Southwestern China. Hematology. 2022;27(1):672–683. doi:10.1080/16078454.2022.2080427
- Xu A, Chen W, Xie W, Wang Y, Ji L. Hemoglobin variants in southern China: results obtained during the measurement of glycated hemoglobin in a large population. Clin Chem Lab Med. 2020;59(1):227–232. doi:10.1515/cclm-2020-0767
- Brennan SO, Tauro GP, Melrose W, Carrell RW. Haemoglobin Port Phillip alpha91 (FG3) Leu replaced by Pro, a new unstable haemoglobin. FEBS Lett. 1977;81(1):115–117. doi: 10.1016/0014-5793(77)80940-X.
- Chen Y, Zhang S, Wang C, et al. Effect of high-throughput sequencing for the prevention and control of thalassemia. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2020;37(6):645–649. doi:10.3760/cma.j.issn.1003-9406.2020.06.012
- Du L, Bao X, Qin D, et al. Compounded with hemoglobin Port Phillip and -alpha(4.2) or --(SEA) deletions were identified in Chinese population. Mol Genet Genomic Med. 2021;9(9):e1699. doi:10.1002/mgg3.1699
- Tan M, Bai Y, Zhang X, et al. Early genetic screening uncovered a high prevalence of thalassemia among 18309 neonates in Guizhou, China. Clin Genet. 2021;99(5):704–712. doi:10.1111/cge.13923
- Baruah A, Baruah MK. Phenotypic diversity and clinico-hematological profile of Hb E-Beta thalassemic children. Indian J Hematol Blood Transfuse. 2020;36:(1):117–122. doi:10.1007/s12288-019-01150-5
- Chuansumrit A, Sirachainan N, Kitpoka P, et al. The Effect of blood transfusion on growth of patients with Hb E/β-Thalassemia. Hemoglobin. 2019;43(4–5):264–272. doi:10.1080/03630269.2019.1692863
- Pornprasert S, Panyasai S, Kongthai K. Comparison of capillary electrophoregram among heterozygous Hb Hope, Hb Hope/alpha-thalassemia-1 SEA type deletion and Hb Hope/beta(0)-thalassemia. Clin Chem Lab Med. 2012;50(9):1625–1629. doi:10.1515/cclm-2012-0016
- Chen YP, Wu P, Wang H, et al. A Rare Case of Abnormal Hemoglobin Variant Hb Mizuho: [HBB: c.206T > C β 68(E12) Leu-Pro]: a First Report in the Chinese Population. Hemoglobin. 2023;28:1–5 doi:10.1080/03630269.2023.2231851