
Abstract
Hypertransfusion regimens for thalassemic patients revolutionized the management of severe thalassemia; transforming a disease which previously led to early infant death into a chronic condition. The devastating effect of the accrued iron from chronic blood transfusions necessitates a more finely tuned approach to limit the complications of the disease, as well as its treatment. A comprehensive approach including carefully tailored transfusion protocol, continuous monitoring and assessment of total body iron levels, and iron chelation are currently the mainstay in treating iron overload. There are also indications for ancillary treatments, such as splenectomy and fetal hemoglobin induction. The main cause of death in iron overload continues to be related to cardiac complications. However, since the widespread use of iron chelation started in the 1970s, there has been a general improvement in survival in these patients.
Introduction
The introduction of hypertransfusion regimens in the 1960s for thalassemic patients revolutionized the management of severe thalassemias; transforming a disease which previously led to early infant death into a chronic condition. Although chronic blood transfusion regimens have added decades to the lives of patients, clinicians are now faced with increasingly complicated management challenges. The devastating effect of the accrued iron from chronic blood transfusions necessitates a more finely tuned approach to limit the complications of the disease, as well as its treatment. Survival in transfusion-dependent thalassemia patients can be improved with proper understanding of the pathophysiology of thalassemia and iron toxicity, comprehensive transfusion protocols, accurate measurements of total body iron, and employment of strategies to reduce iron burden.
Pathogenesis
In the post-uterine life, two types of hemoglobins predominate, hemoglobin A1 and hemoglobin A2. Both of these are made up of four tetramers, either two α globulins and two β globulins (hemoglobin A1) or two α globulins and two δ globulins (hemoglobin A2). In addition, a small percentage of fetal hemoglobin (HbF) is present and is made up of two α globulins and two γ globulins.
β-Thalassemia
The synthesis of α globulin and β globulin is tightly regulated, such that the ratio is close to 1:1. In β thalassemia there is a decrease or absence of the β globulin leading to a relative increase in the number of α globulins. The excess and unpaired α globulins are less soluble and form precipitates that disrupt the normal function of red blood cells leading to a variety of clinical manifestations.
β-thalassemia is broadly clustered into three groups based on degree of clinical severity.
β-thalassemia Minor
β-thalassemia minor, also known as “β-thalassemia carrier” or “β-thalassemia trait”, is the asymptomatic form of β-thalassemia. However, on rare occasions, some patients require blood transfusion during pregnancy due to severe anemia. Often, they are misdiagnosed as having iron deficiency, which is more common, until the diagnosis comes into question after a lack of response to iron supplementation. Once a carrier state is discovered, these patients should seek genetic counseling before having children.
β-thalassemia Intermedia
β-thalassemia intermedia usually presents in late childhood and requires infrequent to no transfusions. These patients still require proactive treatment to prevent serious complications, such as splenomegaly, extramedullary hematopoiesis, which may cause skeletal deformities and hypercoagulable states, which could lead to thrombotic events. Close monitoring, especially during periods of heightened stress, such as surgery, pregnancy, and rapid growth, is essential as this may exacerbate the condition to the extent that the implementation of a transient transfusion regimen may become necessary. Many clinicians advocate the use of early blood transfusion since it allows for normal growth and decreases the risk of developing extramedullary hematopoiesis.Citation1 At the moment, no evidence-based guidelines have been developed for treatment of thalassemia intermedia and treatment is performed on an individualized basis, with the objective of limiting its long-term complications.
Interestingly, even without receiving regular blood transfusions, iron overload is still common among patients with thalassemia intermedia. The body compensates for the chronic state of anemia by increasing intestinal absorption of iron. Although the iron accumulates more slowly, it may eventually reach levels similar to those of transfusion- dependent patients. Therefore, it is prudent to regularly monitor the iron levels and if elevated, especially in the liver and heart, iron chelation therapy should be initiated.Citation2,Citation3 Since the iron overload is more physiological, rather than iatrogenic, serum ferritin does not accurately reflect the true body iron. Magnetic resonance imaging (MRI) appears to be superior in determining iron levels in these patients.Citation4
When patients with β-thalassemia intermedia reach old age, the accumulation of the adverse effects of decades of chronic anemia may lead to serious end organ damage such as cardiopulmonary compromise or signs of progressive bone marrow expansion (pathologic fractures, nerve impingement). To avoid these complications, initiation of chronic blood transfusion during this period may be necessary. The presence of non-transfusional iron overload makes the management of iron toxicity in these patients particularly challenging.
β-thalassemia Major
β-thalassemia major, also known as “Cooley’s anemia” and “Mediterranean anemia” has a much earlier onset (6–24 months) and these patients require regular transfusions. Patients usually present with failure to thrive, progressive paleness, feeding problems, diarrhea, recurrent fever, or enlarged abdomen secondary to liver or spleen enlargement. Without treatment, the condition is further characterized by growth retardation, pallor, jaundice, poor musculature, genu valgum, hepatosplenomegaly, leg ulcers, development of masses from extramedullary hematopoiesis and skeletal changes. Mongoloid facies such as bossing, skull, depression of nasal bridge is also common. Since chronic blood transfusion protocols are applied primarily to patients with β-thalassemia major, the information presented in this review article will be primarily focused on patients with this subset of thalassemia.
Alpha-thalassemias
In α-thalassemia, there is a decrease or absence in the production of α globulin. The severity of α-thalassemia encompasses both ends of the spectrum. The most severe form, hemoglobin Barts, is incompatible with intrauterine life and the mildest forms, α-thalassemia silent carrier and α-thalassemia minor are essentially asymptomatic and do not require management. The only α-thalassemia that may require chronic treatment is hemoglobin H disease.Citation5 Although it is commonly classified under non-transfusion dependent thalassemias,Citation6 these patients may end up requiring blood transfusions in the second decade of life. Regardless of chronic blood transfusions, they may still require iron chelation due to iron overload from increased intestinal iron absorption, analogous to β-thalassemia intermedia, and should be treated similarly.
Transfusion protocol
The current practice guidelines for management of β-thalassemia major patients involve lifelong, regular blood transfusions. The primary goals of blood transfusion therapy include:
Maintaining optimal viability and function of the red blood cells to ensure sufficient oxygen transport to the tissues and organs to avoid the complications of chronic anemia and to suppress ineffective erythropoiesis.
Achieving an appropriate hemoglobin level while avoiding iron overload.
Avoiding adverse reactions of blood transfusion therapy with appropriate red cell antigen typing, cross matching, and screening for new antibody production.Citation7
Chronic blood transfusions prevent most of the comorbidities of β-thalassemia major including growth, skeletal, and neurological complications. The decision to start chronic blood transfusion therapy requires input from the patient, family, and the medical team. Criteria for initiation of blood transfusion does not depend solely on the presence of anemia, but also on the inability to compensate for the anemia impacting the patient’s quality of life and on the increasing symptoms of ineffective erythropoiesis.
Starting regular transfusions is indicated when initial hemoglobin level drops below 6 g/dL. However, anemia caused by sepsis or viral infection must be ruled out first by withholding transfusions and monitoring hemoglobin levels weekly. Thus, regular transfusions should be inaugurated with the goal to maintain the pre-transfusion hemoglobin levels ≥9 g/dL in order to suppress extramedullary hematopoiesis, therefore minimizing skeletal changes, growth impairment, and splenomegaly.
The baseline laboratory investigations required before initiating blood transfusion include obtaining an extended red cell phenotype to reduce the risk of developing alloantibodies. In addition, antibodies to hepatitis B and C, and HIV should be acquired along with serum bilirubin, transaminase, and serum ferritin levels.
The blood product that is preferably administered is packed red blood cells depleted of leukocytes that are matched with the patient’s red cell antigen phenotype with at least D, C, c, E, e, and Kell. Whole blood is unsuitable for chronic transfusions as the chance for non-hemolytic transfusion reaction to develop is common.
Blood transfusions are administered every 3 to 4 weeks at a rate of 15–20 mL/kg to maintain the pre-transfusion hemoglobin level between 9 to 10 g/dL and enough to suppress extramedullary hematopoiesis. Post-transfusion hemoglobin level should not exceed 14 g/dL. On the other hand, in patients with severe anemia with a hemoglobin level below 5 g/dL or who have cardiac insufficiency, the rate of transfusion should be reduced to circumvent fluid overload, thus, diuretic usage may be necessary in some patients. Patients with cardiac compromise need to maintain a higher pre-transfusion hemoglobin level, between 10 to 12 g/dL.Citation8
It is important that patients are monitored for the development of any hemolytic reactions developing during transfusion. Febrile and allergic reactions respond well to acetaminophen and diphenhydramine before administering future transfusions. Those that develop allergic reactions must be given washed packed red blood cell units.
Since iron overload is a critical issue of regular blood transfusions, it is very important to monitor the body’s iron load. This includes obtaining monthly serum ferritin levels, monitoring growth charts in pediatric patients, looking for any signs of endocrinopathy, annual T2* MRI done from the age of 7 years, and an annual electrocardiogram to detect any arrhythmias in adolescents. To rule out any adverse effects of iron chelators an audiogram and a vision test is done yearly from the ages of 5 and 10 years respectively.
Assessment of body iron
Since β-thalassemia major patients require frequent blood transfusions to overcome the effects of anemia, iron overload is a common life-threatening complication. Therefore, it is crucial that reliable methods are available to monitor the iron status in transfusion-dependent patients.
Today, several techniques are available for evaluating iron overload, but some are more reliable than others. These include serum markers such as serum iron, ferritin, and total iron binding capacity. However, studies have found that serum ferritin values correlate poorly with hepatic iron concentrationCitation9 and may give a false estimate of the total body iron. Also, serum ferritin may be influenced by a number of other factors, such as inflammatory statesCitation10 and hepatitis, which may show abnormally high serum ferritin levels, and vitamin C deficiency, which may show abnormally low serum ferritin levels.Citation9 In addition, serum ferritin is not useful in predicting iron overload-related complications. Despite some patients having low ferritin levels for decades, they may still experience heart diseases as they age.Citation11 On the other hand, serial serum ferritin values are useful in monitoring iron chelator responsiveness as this method is easy, cheap, and easily accessible.Citation10
Since serum ferritin levels underestimate the body’s total iron, some centers select more invasive procedures to assess iron overload in transfusion-dependent patients. Since the liver is the predominant iron storage organ, the liver iron content (LIC) correlates well with the body’s total iron status. Unfortunately, determination of the LIC requires a liver biopsy, which is more expensive, invasive, and carries serious risks, such as hemorrhage.Citation10
To overcome the drawbacks of liver biopsy, several non-invasive techniques have arisen to estimate tissue iron concentration. These include the superconducting quantum interference device (SQUID), quantitative computed tomography (CT), and MRI.Citation10
Among the first non invasive techniques used to measure tissue’s iron load was SQUID.Citation11 SQUID utilizes very low power magnetic fields with sensitive detectors placed over the liver to measure the magnetic flux created from the interference of the patient’s body iron with the field.Citation12 Although the test has shown linear correlations with LIC measured through liver biopsy, SQUID tends to underestimate LIC values. Additionally, its use is restricted due to limited availability and high costs.Citation9
CT is a well-tolerated, and relatively inexpensive alternative for determining LIC. A non-contrast CT in patients with hepatic hemochromatosis shows a diffuse increase in liver attenuation (usually >75 Hounsfield units). Consequently, the liver vasculature appears more prominent because of the increased contrast between the vessels and the highly attenuated liver as shown in .
Figure 1 Non-contrast CT scans.
Abbreviation: CT, computed tomography.
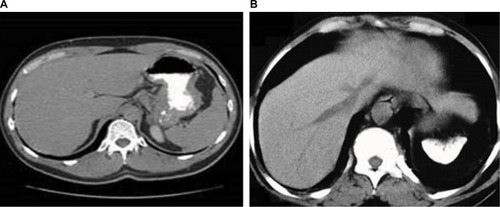
Though CT provides a closer correlation with LIC than serum ferritin levels,Citation13 this technique has been limited by poor sensitivity in patients with low iron levels and increased exposure to ionizing radiation.Citation11
Finally, MRI is the most accurate and widely available tool in detecting and screening for iron overload in endocrine organs, the liver, and the heart. It is currently the preferred imaging modality because of its several advantages including a low rate of variability between measurements and a high sensitivity.Citation10 It has been integrated in the standard of care in not only transfusion-dependent thalassemic patients, but in all chronic transfusion-dependent anemic patients.
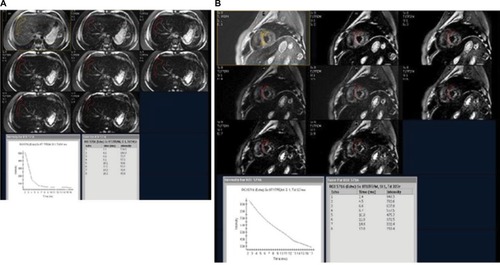
To measure iron concentration in a particular tissue, the MRI transmits a stimulus that excites the water’s protons in the tissue. As the protons relax back to their steady state, they send out microwaves to the MRI scanner which are interpreted into an image. The magnetic environment in non-iron overloaded tissues is homogenous, giving a brighter image and the signals last longer (). On the other hand, in iron overloaded tissues, iron acts like a magnet disrupting the coherence between the protons and darkening the image quicker ().Citation10 This darkening process which is proportional to tissue iron concentration is described by a “half-life” as T2* with a gradient echo imaging and T2 which is provided by a spin echo. The rate of darkening is defined as R2*, which is a reciprocal of T2*. In summary, the greater the tissue iron load, the shorter the signal half-life, thus giving a smaller T2 and T2* on the MRI.Citation11
Figure 2 Normal axial T2* of liver of a 21-year old male patient with a known case of β-thalassemia on regular blood transfusions.
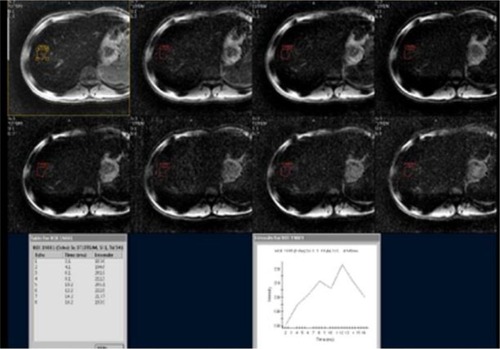
Figure 3 Axial T2* of liver and heart of a 24-year old female patient with a known case of β-thalassemia major on chronic blood transfusions.
Research is still underway to find more accurate and less invasive methods of measuring total body iron. A recent study has shown that total body iron can be measured and monitored by applying a Perl’s Prussian blue stain to exfoliative buccal cells of β-thalassemic major patients. These results were considered significant, however, further research is required to support these findings.Citation14
It is important that chronically transfused patients have serial follow-ups to determine whether the patient is responding to chelation therapy properly in order to avoid the life-threatening complications of iron overload on the tissues, however, the technique needs to be easy, sensitive, cheap, and widely available. Thus, several institutes have adopted the MRI as an effective technique to serially monitor iron overload in chronic transfusion patients.
Iron toxicity
Each unit of transfused blood contains 200 to 250 mg of elemental iron. The body has not developed adequate mechanisms to excrete such quantities of iron. Therefore, the development of iron overload in patients receiving chronic blood transfusion is inevitable. After the iron storage ability of the reticuloendothelial cells is saturated, the iron spills into blood where it normally binds to transferrin. Once transferrin is oversaturated the level of non-transferrin bound iron steadily increases. The unbound iron begins to accumulate in various organs, such as the heart, liver, pancreas, gonads, and pituitary, and causes the catalysis of injurious compounds, such as free radicals. These free radicals begin to damage cells leading to fibrosis or organ dysfunction.Citation15
When the unbound iron is taken up by cardiac tissue, the steady accumulation of oxygen metabolites and other toxin formed from reactions catalyzed by the iron ultimately destroy the tissue. Eventually, there is development of left ventricular hypertrophy and conduction disturbances in late childhood. If untreated, these patients may develop refractory congestive heart failure by their mid-teens.Citation16
Histological studies have shown deposition of iron in pituitary gland leading to hypogonadotropic hypogonadism and growth hormone deficiency, despite iron chelation therapy.Citation17–Citation19
Reducing iron burden
Currently the mainstay of managing transfusion-related iron toxicity is through the use of iron chelators, however, other strategies to limit iron burden are also employed.
Iron chelators
Iron chelators work by neutralizing unbound iron and removing excess iron in tissues. Iron has six active sites and to achieve complete inactivation of a single atom of iron, all six sites must be bound by the chelator. Hexidentate chelators, such as deferoxamine, can bind to all six sites, while tridentate chelators such as deferasirox can bind to three sites and bidentate chelators, such as deferiprone, can bind to only two sites. Therefore, a hexidentate chelator binds at a 1:1 ratio, while a tridentate chelator binds at a 2:1 ratio, and a bidentate chelator binds at a 3:1 ratio. The non-hexidentate chelators, deferasirox and deferiprone, will produce a mixture of irons with variable levels of inactive sites and only a minority of those irons will be completely inactive.
The use of iron chelating agents has significantly increased the survival rates and decreased morbidity related to organ system toxicity. Ample numbers of studies have illustrated that iron chelators are effective in ceasing and often reversing liver and cardiac dysfunction attributed to iron overload.Citation16,Citation20 Chelation therapy has also been shown to reduce the risk of diabetes mellitus and glucose intoleranceCitation21 and, if used early, allows most patients to reach normal puberty, and increases fertility rates.Citation22
Currently, there are a variety of iron chelators, each with different properties, to choose from. An ideal chelator should bind to a large amount of ferric iron at low doses and toxicity, while having a low affinity for ferrous iron and other metals. Since iron often enters the cell during iron overload, intracellular penetration is also important. For better compliance, the drug must have good oral tolerability, affordability, and availability.
Deferoxamine
Deferoxamine is the first iron chelator to be used in treatment of iron toxicity. Since deferoxamine has been around for over 50 years, there has been significant clinical experience. Due to its established efficacy it continues to be the standard iron chelator used. In a 10-year New England Journal of Medicine study published in 1994, all pediatric patients who were able to achieve controlled serum ferritin levels with 12-hour subcutaneous deferoxamine infusion survived without cardiac disease.Citation23 Other studies have confirmed this, showing a significant decline in mortality and heart failure.Citation23,Citation24
Deferoxamine is capable of reversing hepatic iron overload. In the liver, deferoxamine is internalized by hepatic parenchymal cells where it binds with intracellular iron and is excreted in bile. Studies in pediatric groups showed that intermittent deferoxamine infusion decreased iron storage in the liver and prevented hepatic fibrosis.Citation20 However, patients with concurrent chronic hepatitis may require dose adjustments.Citation25
Deferoxamine is also able to remove iron from cardiac cells. Iron released by macrophages is immediately chelated and excreted in urine. Within a few hours of administration, the urinary iron concentrations increase significantly in a dose-dependent fashion.Citation26 Each nightly infusion should have a loss of 20 to 50 mg/day of iron in urine and stool, which should be effective at controlling iron levels, if the patient is receiving under four units of packed red blood cells per month.Citation27
The disadvantages of deferoxamine are related to its short half-life and parenteral requirements. Patients usually require nightly continuous subcutaneous infusions 5–7 days/week of 2 g deferoxamine via a pump. This is not only difficult on the patient, but is labor intensive and necessitates the installation of semi-permanent intravenous access, which is associated with additional complications.Citation28 Subcutaneous injections are an alternative, but are painful. Both avenues increase the likelihood of non-compliance with therapy, which may be as low as 50% in children and adolescent populations. Patients who are not fully compliant with deferoxamine treatment have substantially greater morbidity and mortality as well as increased costs.Citation29 Therefore, many investigators have pushed for oral agents.
Some reports have suggested that early and aggressive deferoxamine administration may adversely affect skeletal maturation and result in growth retardation.Citation30 However, there is also evidence that early chelation may delay cardiac symptoms and prolong survival, therefore some centers choose to begin deferoxamine infusions around 5 years of age.Citation31
Overall, deferoxamine is well tolerated by most patients. Many patients experience minor local irritation at the injection site, which can be controlled by reducing the dosage or the rate of infusion. Therefore, frequent monitoring for complications is important. These complications include:Citation32
Ototoxicity, which is often irreversible, can occur when the ratio of deferoxamine (mg/kg) to serum ferritin (μg/L) is greater than 0.025.Citation33 Therefore, audiology exam should be carried out before treatment. Another screening should be performed after 6 months and another formal audiogram should be administered 12 months into treatment. It is important to detect any hearing loss early because decreasing the dosage can prevent hearing loss.Citation34
Ocular toxicity may present as night blindness, blurred vision and decreased visual acuity, impaired color vision or cataract. An ophthalmologist should evaluate children every 6 months and adults annually as well as routinely asking about vision changes during visits.
Retarded growth and skeletal changes.
Infections, such as Yersinia enterocolitica and other pathogens.
Pulmonary and neurological disease is associated with doses greater than 60 mg/kg.
Renal function may be affected, therefore creatinine/blood urea nitrogen should be monitored every 3 months. Decrease dosage if serum creatinine increases by 30% to 50% from baseline and discontinue temporarily if the serum creatinine rises by greater than 50% from baseline or if the protein/creatinine ratio is greater than 0.6 mg/mg.Citation35
Liver function should be monitored every 3 months and chelators should be stopped if ALT is greater than five times the upper limit.
Many serious complications can be avoided by utilizing doses less than 2.5 g with each infusion. Once serious complications develop, they can generally be controlled by discontinuing the drug or decreasing its dosage. These complications, coupled with poor compliance as a result of inconvenient parenteral administration, has limited the effectiveness of this treatment as a chelator. The need for a better oral iron chelator was readily apparent and so the development of these alternatives was undertaken.
Deferiprone
Deferiprone was introduced in the 1980s and was the first oral iron chelator to be used and works by forming stable complexes with plasma iron that can be excreted through urine.Citation36 Clinical experience over the past decades have shown deferiprone to be effective in increasing iron excretion, however, deferoxamine still appears to be slightly more effective. This has been attributed to fecal iron excretion as a result of deferoxamine.Citation37 A 2006 randomized controlled trial concluded deferiprone to more effective in improving myocardial siderosis than deferoxamine.Citation38 However, a follow-up meta-analysis in 2008, failed to draw the same conclusion, rather, they found that there is no significant difference between deferoxamine and deferiprone in reducing iron concentration.Citation39 For the moment, it seems more studies and longer follow-ups are required before a definitive conclusion can be reached.
Like deferoxamine, deferiprone has a short half-life which requires multiple daily dosing.Citation40 The recommended dosage is between 75–100 mg/kg/day divided into three doses. The most serious adverse effect of deferiprone is reversible agranulocytosis, usually within the first year, but reported up to 19 months.Citation36,Citation41 This effect tends to recur with continuation of treatment. At the moment, it is not clear whether this is an idiosyncratic effect or dose-related. Due to this reaction, weekly monitoring of white blood cells in the first year and then 2 weeks thereafter is recommended to detect early neutropenia or agranulocytosis. Discontinue if the neutrophil levels drop below 1,500 cell/μL.
Gastrointestinal tract symptoms, such as nausea, vomiting, and abdominal pain have also been reported in up to 33% of patients, however, they are typically mild.Citation42 Zinc deficiency is a rare occurrence, usually seen in diabetic patients. Hence, zinc levels should be checked every 3 to 6 months in diabetic patients. Provide zinc supplements if zinc is determined to be low.
Deferiprone may also lead to elevated liver enzymes, which may be attributed to other associated conditions such as hepatotoxicity. Chelation therapy should be halted if ALT is five times the upper limit of normal or if transaminases are persistently two times the upper limit of normal.Citation43
Although compliance is substantially higher with deferiprone than deferoxamine,Citation29 the efficacy and safety of deferiprone still have not been established, which has precluded deferiprone from routine use.Citation44 Many researchers have begun to study whether a combination treatment of deferoxamine and deferiprone is more effective than monotherapy. A 2007 randomized, placebo-controlled, double-blind trial comparing combination versus monotherapy concluded combination therapy to be more beneficial in improving cardiac function and reducing cardiac iron burden.Citation45 However, a subsequent 2013 meta-analysis did not find enough evidence to warrant a recommendation to begin combination therapy in all patients. Their recommendation maintained that deferiprone should be reserved for patients in whom deferoxamine is contraindicated or inadequate.Citation46
Deferasirox
Deferasirox is the latest oral iron chelator and has undergone extensive studies including a Phase 3 clinical trial. The trial indicated that daily administration of 30 mg/kg/day could decrease LIC similar to deferoxamine at 50 mg/kg/day. The long half-life of deferasirox (8–16 hours) allows for once-daily dosing, which is one of the major advantages of this drug.Citation47
The CORDELIA study, was a prospective, randomized study that compared the usage of deferasirox versus deferoxamine for myocardial iron removal in β-thalassemic major patients with myocardial siderosis, but no cardiac malfunction. This study demonstrated that there is no superiority of deferoxamine compared to deferasirox for myocardial iron removal.Citation48
During the trial, only mild adverse effects, such as abdominal pain, back pain, and skin rash were reported. However, now post-marketing reports of acute renal failure, cytopenias, hepatic failure, and gastrointestinal hemorrhage, often fatal, have surfaced. It has already been noted that these reactions were more frequent in patients with advanced age, high-risk myelodysplastic syndromes, underlying renal or hepatic impairment or low platelet counts.Citation49 Many studies have also reported the presence of acquired proximal renal tubular dysfunction.Citation50,Citation51
Consequently, it is important to routinely perform liver function tests and renal function tests. It is generally advised to discontinue the chelator if serum creatinine is greater than two times normal or increases by 50% from baseline or if the protein-to-creatinine ratio is >0.6 mg/mg. It should also be discontinued if the serum ALT is greater than five times the upper limit.
Choosing the chelator
No iron chelating agent is considered “gold standard” and clinicians differ on the optimum approach to initiating iron chelation therapy in patients with transfusion-dependent thalassemia. Moderate doses of deferasirox can maintain iron balance in a majority of patients on blood transfusion. It tends to be preferred over deferoxamine because of greater compliance, especially since poor compliance is associated with poor survival.Citation29 However, it must be explained to patients that more data on long-term efficacy have been established for deferoxamine.
The choice of specific iron chelator is based on factors other than the efficacy of the drug. Other factors such as age, patient preference, comorbidities, and drug side effects will ultimately determine which chelator is used (). Patients with high iron intake may require higher dose deferasirox or combination regimens with deferoxamine. Deferiprone remains as a second line drug and plays a larger role if there is significant cardiac toxicity from iron overload.
Table 1 Comparison of iron chelators
In certain cases, monotherapy may be insufficient to achieve treatment goals. At this point the clinicians should either increase the current dose of the drug or switch to a different chelator. However, if treatment goals are still not achieved, then the clinician may opt to initiate combination therapy. Combination chelators tend to be more effective than monotherapy because chelators utilize different mechanisms for removing iron. Combination therapy has been shown to be useful for patients with mild-to-moderate hepatic and cardiac iron overload, as well as severe degrees of cardiac siderosis and impaired left ventricular function.Citation52,Citation53
When to start iron chelators
Starting a vigorous iron chelation therapy earlier has been associated with significant improvement in survival.Citation54 The general rule is to begin iron chelation treatment when the following parameters are present:Citation55
After transfusion of 100 mL/kg packed red blood cells (10–20 transfusions).
Ferritin levels above 1,000 ng/mL.
Liver iron concentration greater than 3 mg/g dry weight of liver as measured by liver biopsy or MRI.
Dosing
No set dosing has been specified and most clinicians tailor dosing to achieve particular laboratory parameters. Dosing is steadily increased until serum ferritin levels are <1,000 μg/L, LIC <7 mg Fe/g dry weight, and cardiac T2* by MRI >20 millisecond. It can take anywhere from 3 to 5 years to reduce serum ferritin levels below 1,000 ng/mL with deferoxamine alone. Once serum ferritin falls below 300 to 500 μg/L and/or LIC falls below 3 mg/g dry weight, chelation therapy can be reduced or discontinued.Citation45 Dosage should be increased if ferritin and LIC levels increase on average over a period of 6 months. Keep in mind not to exceed levels which may lead to complications, as discussed earlier.
Intensification of treatment with increased dosage, frequency or combination chelators should be considered when: LIC >15 mg Fe/g, serum ferritin >2,500, cardiac T2* MRI <15 milliseconds or a fall in left ventricular ejection fraction due to siderosis, cardiac failure or arrhythmia. Often patients on deferoxamine are noncompliant and therefore these patients should be switched to deferiprone.Citation52,Citation53
Improvements in survival
Survival of patients with β-thalassemia major is influenced by several factors ranging from age, education, ferritin levels, complications, and many more. However, over the past few decades a steady increase in survival has been documented. This was highlighted in a 2005 study where survival probabilities estimated by the Kaplan–Meier method displayed that survival was higher for patients born after 1975 compared to those born before.Citation56
Despite these advances the most common cause of death remains cardiac complications (68%) due to iron overload.Citation24 Therefore, control of iron overload remains the best prognostic factor. Despite the problems with using serum ferritin as a measure of total body iron, as mentioned earlier, results from several studies including those carried out in Iran and Italy, have suggested that serum ferritin levels affect the longevity of thalassemic patients. These studies emphasize that serum ferritin levels >2,500 ng/mL are associated with reduced survival.Citation24,Citation57
A study carried out in 1991 divided patients into two groups. Group 1 followed a low-transfusion protocol with no chelation, whereas Group 2 followed a hypertransfusion regimen with deferoxamine. Group 1 had an estimated median age of survival of 17.4 years compared to 31 years in Group 2. This study concludes deferoxamine treatment, when used in amounts proportionate to the iron burden, can delay cardiac complications and prolong survival.Citation58
Splenectomy
Splenectomies allow red blood cells to live longer and therefore the patient can maintain higher levels of hemoglobin for longer periods and decrease their requirement for transfusions. Splenectomies are performed when the transfusion requirements begin to exceed 200 mL/kg or the transfusion requirements increase by 50% over a 1-year period. Unfortunately, removal of the spleen does have other complications, such as increased risk of infection and thrombosis, therefore the benefits and risk of splenectomies must be carefully weighed before initiating this course of treatment.Citation31 It is advisable to delay splenectomy until after the age of 5 years, when proper immunizations can be dispensed to reduce the increased risk of post-splenectomy infection.Citation59
Induction of HbF
As mentioned earlier, HbF does not have a β globulin, therefore by inducing more HbF, it may serve to correct the excess α chain imbalance. Normally, HbF makes up less than 3% of the total hemoglobin, however, certain cytotoxic drugs and cancer chemotherapy agents are able to increase the production of HbF, such as decitabine, butyrate, and hydroxyurea.
Many studies have shown a decrease in transfusion requirements or complete transfusion independence, after the administration of hydroxyurea.Citation60,Citation61 Therefore, HbF induction is an effective strategy to reduce iron burden by decreasing the frequency of transfusions. Unfortunately, hydroxyurea is unable to induce the production of HbF in all patients and treatment responses are highly variable. Currently, no randomized controlled study has been done, so more studies are required before stronger conclusions about the efficacy of hydroxyurea can be drawn.
Although no ideal HbF inducer exists, the therapeutic benefits of HbF induction is undeniable. Currently, many clinicians administer fetal globulin inducers on a trial basis, especially to patients with high transfusion requirements and patients experiencing complications from blood transfusions, such as alloimmunization.
Conclusion
Thalassemia syndromes result from mutations in the α or β globulin that disrupts the normal globulin ratio. The presentation of these syndromes is clinically diverse, ranging from a carrier state to a life-threatening hemolytic anemia requiring chronic blood transfusion. Managing patients with transfusion-dependent β-thalassemia major still remains a major challenge to clinicians. Despite an effective transfusion protocol, transfusion hemosiderosis is a major cause of mortality in these patients. However, with a proper, systematic approach from the medical team and regular follow-ups, the complications of iron overload can be detected early and reduced. Proper management requires regular and accurate monitoring of total body iron and timely administration of treatment modalities to reduce iron burden. Fortunately, the emergence of effective iron chelators, although cumbersome to administer, has substantially improved the quality of life and care for these patients.
Disclosure
The authors declare that there are no conflicts of interest regarding the publication of this paper.
References
- TaherATMusallamKMCappelliniMDWeatherallDJOptimal management of β thalassaemia intermediaBr J Haematol2011152551252321250971
- TaherATViprakasitVMusallamKMCappelliniMDTreating iron overload in patients with non-transfusion-dependent thalassemiaAm J Hematol201388540941523475638
- LadisVBerdousiHGotsisEKattamisADeferasirox administration for the treatment of non-transfusional iron overload in patients with thalassaemia intermediaBr J Haematol2010151550450820950401
- TonySDaarSElshinawyMAl-ZadjalySAl-KhaboriMWaliYT2* MRI in regularly transfused children with thalassemia intermedia: serum ferritin does not reflect liver iron storesPediatr Hematol Oncol201229657958422839111
- ChuiDHFucharoenSChanVHemoglobin H disease: not necessarily a benign disorderBlood2003101379180012393486
- MusallamKMRivellaSVichinskyERachmilewitzEANon-transfusion-dependent thalassemiasHaematologica201398683384423729725
- QariMHWaliYAlbagshiMHRegional consensus opinion for the management of Beta thalassemia major in the Arabian Gulf areaOrphanet J Rare Dis2013814324044606
- Northern California Comprehensive Thalassemia CenterStandard-of-Care Clinical Practice Guidelines (2012) Available from: http://thalassemia.com/treatment-guidelines-4.aspx#gsc.tab=0Accessed June 27, 2016
- PrabhuRPrabhuVPrabhuRSIron overload in beta thalassemia: a reviewJ Biosci Tech2009112031
- WoodJCMagnetic resonance imaging measurement of iron overloadCurr Opin Hematol200714318319017414205
- WoodJCDiagnosis and management of transfusion iron overload: the role of imagingAm J Hematol20078212 Suppl1132113517963249
- ShethSSQUID biosusceptometry in the measurement of hepatic ironPediatr Radiol20033637337712768253
- ChapmanRWWilliamsGBydderGDickRSherlockSKreelLComputed tomography for determining liver iron content in primary haemochromatosisBr Med J198028062124404427370525
- BhatAAParwaniRNWanjariSPDemonstration of iron in exfoliated buccal cells of β-thalassemia major patientsJ Cytol201330316917324130408
- BaconBRBrittonRSThe pathology of hepatic iron overload: a free radical-mediated processHepatology19901111271372153094
- EngleMAErlandsonMSmithCHLate cardiac complications of chronic, severe, refractory anemia with hemochromatosisCirculation19643069870514226168
- GrundyRGWoodsKASavageMOEvansJPRelationship of endocrinopathy to iron chelation status in young patients with thalassaemia majorArch Dis Child19947121281327944532
- KwanEYLeeACLiAMA cross-sectional study of growth, puberty and endocrine function in patients with thalassaemia major in Hong KongJ Paediatr Child Health199531283877794630
- De SanctisVGrowth and puberty and its management in thalassemiaHorm Res200258Suppl 1727912373018
- BarryMFlynnDMLetskyEARisdonRALong-term chelation therapy in thalassaemia major: effect on liver iron concentration, liver histology, and clinical progressBr Med J19742590916204821036
- BrittenhamGMGriffithPMNienhuisAWEfficacy of deferoxamine in preventing complications of iron overload in patients with thalassemia majorN Engl J Med199433195675738047080
- Bronspiegel-WeintrobNOlivieriNFTylerBAndrewsDFFreedmanMHHollandFJEffect of age at the start of iron chelation therapy on gonadal function in β-thalassemia majorN Engl J Med1990323117137192388669
- OlivieriNFNathanDGMcMillanJHSurvival in medically treated patient with homozygous β-thalassemiaN Engl J Med199433195745788047081
- Borgna-PignattiCRugolottoSDe StefanoPSurvival and complications in patients with thalassemia major treated with transfusion and deferoxamineHaemotologica2004891011871193
- De VirgiliisSCossuPSannaGIron chelation in transfusion-dependent thalassemia with chronic hepatitisActa Haematol198267149566800202
- SmithRSIron excretion in thalassaemia major after administration of chelating agentsBr Med J1962253191577158013989576
- SchaferAIRabinoweSLe BoffMSBridgesKCheronRGDluhyRLong-term efficacy of deferoxamine iron chelation therapy in adults with acquired transfusional iron overloadArch Intern Med19851457121712213925909
- DavisBAPorterJBLong-term outcome of continuous 24-hour deferoxamine infusion via indwelling intravenous catheters in high-risk β-thalassemiaBlood20009541229123610666195
- DeleaTEEdelsbergJSofryginOConsequences and costs of noncompliance with iron chelation therapy in patients with transfusion-dependent thalassemia: a literature reviewTransfusion200747101919192917880620
- De VirgillisSCongiaMFrauFDeferoxamine-induced growth retardation in patients with thalassemia majorJ Pediatr198811346616693171791
- RachmilewitzEAGiardinaPJHow I treat thalassemiaBlood2011118133479348821813448
- CunninghamMJMacklinEANeufieldEJCohnARThalassemia Clinical Research Network. Complications of beta-thalassemia major in North AmericaBlood20041041343914988152
- PorterJBJaswonMSHuehnsEREastCAHazellJWDesferrioxamine ototoxicity: evaluation of risk factors in thalassaemic patients and guidelines for safe dosageBr J Haematol19897334034092605127
- OlivieriNFBuncicJRChewEVisual and auditory neurotoxicity in patients receiving subcutaneous deferoxamine infusionsN Engl J Med1986314148698733485251
- YacobovichJStarkPBarzilai-BirenbaumSAcquired proximal renal tubular dysfunction in β-thalassemia patients treated with deferasiroxJ Pediatr Hematol Oncol201032756456720733517
- PigaARoggeroSSalussoliaIMassanoDSerraMLongoFDeferiproneAnn N Y Acad Sci20101202757820712776
- CappelliniMDMusallamKMTaherATOverview of iron chelation therapy with desferrioxamine and deferiproneHemoglobin200933Suppl 1S58S6920001633
- Borgna-PignattiCCappelliniMDDe StefanoPCardiac morbidity and mortality in deferoxamine-or deferiprone-treated patients with thalassemia majorBlood200610793733373716373663
- MamtaniMKulkarniHInfluence of iron chelators on myocardial iron and cardiac function in transfusion-dependent thalassaemia: a systematic review and meta-analysisBr J Haematol2008141688289018355381
- AndersonLJWestwoodMAHoldenSMyocardial iron clearance during reversal of siderotic cardiomyopathy with intravenous desferrioxamine: a prospective study using T2* cardiovascular magnetic resonanceBr J Haematol2004127334835515491298
- HoffbrandAVCohenAHershkoCRole of deferiprone in chelation therapy for transfusional iron overloadBlood20031021172412637334
- CohenARGalanelloRPigaADipalmaAVulloCTrictaFSafety profile of the oral iron chelator deferiprone: a multicentre studyBr J Haematol2000108230531210691860
- OlivieriNFBrittenhamGMMcLarenCELong-term safety and effectiveness of iron-chelation therapy with deferiprone for thalassemia majorN Engl J Med199833974174239700174
- PippardMJWeatherallDJOral iron chelation therapy for thalassaemia: an uncertain sceneBr J Haematol200011112511091177
- TannerMAGalanelloRDessiCA randomized, placebo- controlled, double-blind trial of the effect of combined therapy with deferoxamine and deferiprone on myocardial iron in thalassemia major using cardiovascular magnetic resonanceCirculation2007115141876188417372174
- FisherSABrunskillSJDoreeCChowdhuryOGoodingSRobertsDJOral deferiprone for iron chelation in people with thalassaemiaCochrane Database Syst Rev20138CD00483923966105
- CappelliniMDCohenAPigaAA Phase 3 study of deferasirox (ICL670), a once daily iron chelator, in patients with beta-thalassemiaBlood200510793455346216352812
- PennellDJPorterJBPigaAA 1-year randomized controlled trial of deferasirox vs deferoxamine for myocardial iron removal in β-thalassemia major (CORDELIA)Blood2014123101447145424385534
- GlicksteinHElRBShvartsmanMCabantchikZIIntracellular labile iron pools as direct targets of iron chelators: a fluorescence study of chelator action in living cellsBlood200510693242325016020512
- Borgna-PignattiCCappelliniMDDe StefanoPCardiac morbidity and mortality in deferoxamine- or deferiprone-treated patients with thalassemia majorBlood200610793733373716373663
- US Food and Drug Administration [homepage on the Internet]Exjade (deferasirox): Boxed Warning. FDA Safety Alerts for Human Medical Products2010 Available from: http://www.fda.gov/Safety/Med-Watch/%20SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm200850.htmAccessed June 28, 2016
- WonkeBWrightCHoffbrandAVCombined therapy with deferiprone and desferrioxamineBr J Haematol199810323613649827905
- De DomenicoIWardDMKaplanJSpecific iron chelators determine the route of ferritin degradationBlood2009114204546455119671920
- HoffbrandAVTaherACappelliniMDHow I treat transfusional iron overloadBlood2012120183657366922919029
- OlivieriNFBerrimanAMTylerBJDavisSAFrancombeWHLiuPPContinuous intravenous administration of deferoxamine in adults with severe iron overloadAm J Hematol199241161631503101
- LadisVChouliarasGBerdousiHKanavakisEKattamisCLongitudinal study of survival and causes of death in patients with thalassemia major in GreeceAnn N Y Acad Sci2005105444545016339695
- RoudbariMSoltani-RadMRoudbariSThe survival analysis of beta thalassemia major patients in South East of IranSaudi Med J20082971031103518626536
- EhlersKHGiardinaPJLesserMLEngleMAHilgartnerMWProlonged survival in patients with beta-thalassemia major treated with deferoxamineJ Pediatr19911184 Pt 15405452007928
- FosburgMNathanDGTreatment of Cooley’s anemiaBlood19907634354442198956
- LoukopoulosDVoskaridouEStamoulakatouAHydroxyurea therapy in thalassemiaAnn N Y Acad Sci19988501201289668534
- ItaliaKYJijinaFFMerchantREffect of hydroxyurea on the transfusion requirements in patients with severe HbE-beta-thalassaemia: a genotypic and phenotypic studyJ Clin Pathol201063214715020154037