
Abstract
The structural and functional conservation of hemoglobin throughout mammals has made the laboratory mouse an exceptionally useful organism in which to study both the protein and the individual globin genes. Early researchers looked to the globin genes as an excellent model in which to examine gene regulation – bountifully expressed and displaying a remarkably consistent pattern of developmental activation and silencing. In parallel with the growth of research into expression of the globin genes, mutations within the β-globin gene were identified as the cause of the β-hemoglobinopathies such as sickle cell disease and β-thalassemia. These lines of enquiry stimulated the development of transgenic mouse models, first carrying individual human globin genes and then substantial human genomic fragments incorporating the multigenic human β-globin locus and regulatory elements. Finally, mice were devised carrying mutant human β-globin loci on genetic backgrounds deficient in the native mouse globins, resulting in phenotypes of sickle cell disease or β-thalassemia. These years of work have generated a group of model animals that display many features of the β-hemoglobinopathies and provided enormous insight into the mechanisms of gene regulation. Substantive differences in the expression of human and mouse globins during development have also come to light, revealing the limitations of the mouse model, but also providing opportunities to further explore the mechanisms of globin gene regulation. In addition, animal models of β-hemoglobinopathies have demonstrated the feasibility of gene therapy for these conditions, now showing success in human clinical trials. Such models remain in use to dissect the molecular events of globin gene regulation and to identify novel treatments based upon the reactivation of developmentally silenced γ-globin. Here, we describe the development of animal models to investigate globin switching and the β-hemoglobinopathies, a field that has paralleled the emergence of modern molecular biology and clinical genetics.
Introduction
The human β-globin locus on chromosome 11 contains the five β-like globin genes, ε, Gγ, Aγ, δ, and β (). Two β-like globin proteins combine with two of the α-like globins (ζ or α) to form the oxygen-carrying hemoglobin (Hb) tetramer of red blood cells (RBCs). Erythropoiesis during mammalian development is characterized by the progressive appearance of distinct populations of erythroid cells expressing stage-specific forms of Hb at defined sites within the embryo. In humans, the earliest (primitive) RBCs are formed in the yolk sac blood islands and contain embryonic Hb (Hb Gower 1; ζ2ε2). At approximately 12 weeks of gestation, definitive erythropoiesis commences, marked by RBCs containing fetal Hb (HbF; α2γ2), replacing the embryonic form. Finally, the expression of fetal γ-globin declines postnatally with a concomitant increase of β-globin synthesis, resulting in the production of adult Hb (HbA; α2β2) ().Citation1,Citation2
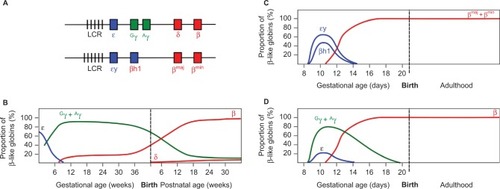
Figure 1 Developmental expression of the β-like globins in humans and in WT and humanized transgenic mice.
Notes: (A) Diagram of the human (upper) and mouse (lower) β-globin loci. Vertical bars represent Dnase I hypersensitive sites in the LCR. Embryonically expressed genes are shown in blue, fetal in green, and adult in red. Switching of the β-like globins in development is shown for (B) human, (C) mice, and (D) human β-like globins in transgenic mice. Values represent the proportion of total β-like globin transcripts detected in erythroid tissue. Note the dual switching events in humans, in contrast to the single mid-gestational switch in WT mice. Note also the mid-gestational switch of human γ- to β-globin expression in transgenic mice.
The β-hemoglobinopathies are caused by mutations in the β-globin gene that lead to the production of aberrant HbS (sickle cell disease, SCD) or, in the case of β-thalassemia, insufficient β-globin synthesis causing ineffective erythropoiesis and life-threatening anemia.Citation3 These conditions manifest clinically in the year following birth, when expression of the fetal γ-globin gene is silenced and replaced by the mutated adult β-globin. Globally, more than 300,000 patients are born each year, representing a substantial demand upon health expenditure.Citation3
The October 13th 1956 issue of Nature contains two articles that represent the early stages of research into the β-hemoglobinopathies. The first, by Ingram,Citation4 describes “A specific chemical difference between the globins of normal human and sickle-cell anemia hemoglobin”. He was following the earlier work of Pauling and Itano,Citation5 who analyzed the charge characteristics of normal and sickle Hb, and proposed a genetic basis for the differences. IngramCitation4 examined the tryptic peptides of normal and sickle Hb and identified a single fragment, unique to HbS, that displayed altered charge characteristics. He correctly attributed this to an alteration in the amino acid sequence of the protein and later identified the Glu-Val substitution responsible.Citation6 On the pages following Ingram’s article, Halbrecht and KlibanskiCitation7 published their paper “Identification of a new normal embryonic hemoglobin”. Electrophoretic mobility was used to compare Hb from blood at 10 and 20 weeks of gestational age with samples from newborn umbilical cord and adult. Most significantly, they reported a drop in the concentration of the new Hb, from 83% at 10 weeks gestation to 52% at 20 weeks. With hindsight, it can be seen that they are describing the first Hb switch, when embryonic Hb (ζ2ε2) is replaced by fetal HbF (α2γ2).
There is a remarkable confluence in the biology underlying these articles that continues to drive research six decades later. The protective effect of HbF in newborns with SCD had been observed prior to the work of Ingram.Citation8 The significance of Hb switching in the context of β-thalassemia began to be recognized with the important work of Fessas and StamatoyannopoulosCitation9 in 1964, describing six families from the Greek Mediterranean region who carried both β-thalassemia and hereditary persistence of fetal Hb (HPFH) traits. HPFH is a nonpathological condition in which the presence of certain genetic polymorphisms leads to incomplete postnatal silencing of the γ-globin gene, resulting in the maintenance of elevated levels of α2γ2 HbF into adulthood. Coinheritance of HPFH with SCD or a β-thalassemia genotype results in milder symptoms due to complementation of the mutant β-globin by the persistent expression of γ-globin. Fessas and StamatoyannopoulosCitation9 observed the mild thalassemia symptoms in individuals displaying elevated HbF and noted that the factor responsible for elevated HPFH “… appears to be very closely linked … to the gene determining structure of the β-chain”. It is likely that they were examining individuals carrying mutations within the β-globin locus itself that lead to HPFH, genetic variants now known to occur in Mediterranean populations.Citation10
The clinical significance of Hb switching, along with the desire to understand the developmental regulation of gene expression, has driven the generation of murine in vivo model systems to study the human fetal-to-adult Hb switch. These models have provided enormous insight into globin switching as an example of developmental gene regulation, although the process is still incompletely understood. In addition, in vivo models of the β-hemoglobinopathies have been created that recapitulate in many aspects the clinical presentation of SCD and β-thalassemia and are valuable tools to investigate novel treatments for these conditions.
Globin switching in the mouse
The laboratory mouse has been the primary model employed for the study of globin switching. Erythropoiesis in the mouse commences in the yolk sac blood islands at ~E7.5, when primitive RBCs appear, expressing the fetal εy and βh1 genes. By mid-gestation (~E12.5), silencing of the fetal genes is in progress and the adult βmaj and βmin are becoming expressed.Citation11 It can therefore be seen that developmental β-like globin gene switching is simpler in the mouse than in humans, occurring only once (). This fact and the brief gestational period of the mouse (21 days) compared to human suggest that biological differences between the species may limit the utility of the mouse as a model. However, over several decades of research, it has become apparent that the underlying mechanisms regulating the β-globin locus are highly conserved across a very wide range of species, and much has been learned from the rodent. Researchers in the field continue to make use of the mouse, recognizing that with careful interpretation, the rodent is an exceptionally valuable resource, furnishing insights into human biology.
Mouse models of human β-globin switching
Transgenic mice created to investigate human β-globin switching began to be reported in the scientific literature in the mid-1980s. The earliest models examined the expression patterns of individual human globin genes in mice.Citation12–Citation14 These studies suggested that the promoter regions immediately upstream of the human globin genes were sufficient to confer developmentally regulated expression in the erythroid lineage, reflective of the pattern of mouse globin switching. Importantly, however, the levels of expression achieved were low, indicating that additional control elements were necessary for physiological levels of transcription.
Subsequently, models were developed employing the human β-globin locus control region (LCR) fused to either an unrelated reporter gene or one of the human globin genes.Citation15–Citation17 These studies demonstrated the capacity of the LCR enhancer to confer high-level erythroid-specific expression of a linked gene. In contrast to the earlier models, the presence of the LCR linked to a single human globin gene apparently overrode the developmental regulation of the transgene, driving the expression of adult human β-globin in embryonic blood.Citation16,Citation18
Subsequent models employed a more physiological arrangement of the transgenic construct, featuring the LCR linked to larger contiguous fragments of the locus, spanning the region from the γ- to the β-globin genes.Citation18 These transgenic loci showed a remarkable recapitulation of developmentally regulated, high-level globin gene expression. Notably, the large size and complexity of the constructs used in these studies permitted a degree of dissection of the role of individual DNase hypersensitive sites within the LCR; partial abrogation of the LCR had a discernible effect upon expression of genes from the transgenic locus.Citation18
The result of these experiments using either individual genes linked to the LCR, or more complex constructs featuring multiple globin genes, was to substantially advance our understanding of the principles underlying regulation of the mammalian β-like globins. The existence of globin switching within birds, mice, and humans, and the evolutionary conservation of the locus structure, suggested that the regulatory mechanisms at work may also possess fundamental similarities between species. Previous work in the chicken led to a “one enhancer–two promoter” hypothesis in which competition between gene promoters for interaction with a single enhancer resulted in preferential expression of the interacting gene.Citation19,Citation20 The expression of developmentally regulated transcription factors was suggested to account for the stage-specific activation of adult or fetal globin genes. The demonstration that age-related regulation of human globin genes in the mouse was dependent upon the presence of both fetal and adult globin genes strongly indicated that this hypothesis held true for mammals. Furthermore, the developmental regulation of the human locus in mice indicated that the regulatory mechanisms responsible were substantially conserved between rodents and primates.
In addition to the conclusions regarding physiological switching, Enver et alCitation16 in 1990 made the particularly insightful suggestion that the competitive switching model explained the phenomenon of deletional HPFH. This group of mutations is characterized by the loss of large segments of the β-globin locus encompassing the δ- and β-genes.Citation21,Citation22 Consequently, in the absence of a competing adult β-globin gene, the intact fetal genes maintain high levels of expression throughout the life of the carrier; indeed, they must do so for the carrier to survive. This situation is mimicked by mice carrying a solitary human Aγ-globin gene linked to the LCR; such animals, lacking an adult β-globin gene in cis, express the fetal transgene at high levels in adult blood.Citation16
The development of yeast and bacterial artificial chromosomes (YACs and BACs, respectively) permitted the propagation and manipulation of large (>100 kb) fragments of DNA. Such technologies made possible the creation of transgenic animals carrying significant stretches of unmodified foreign DNA. Mice harboring YAC fragments of the human β-globin locus further confirmed the developmental regulation of the human globin genes in the rodent, and the presence of all known coding regions and regulatory sequences in their physiological arrangement permitted the results to be interpreted with a greater degree of confidence than those of previous models.Citation23–Citation27 Also, the presence of HPFH point mutations within the human locus delivered as a YAC resulted in transgenic mice that recapitulated the delayed globin switching observed in humans.Citation28
The most significant outcome of these studies was the clear demonstration that, in the mouse, expression of the human γ-globin genes commences in the yolk sac with the embryonic εy and βh1-globin genes, rather than in the fetal stage of definitive erythropoiesis as in humans. Additionally, the expression of transgenic human γ-globin continues beyond the time point at which murine εy and βh1 are silenced (). It should also be noted that the expression of the human β-globin gene from BAC and YAC transgenic loci in the mouse does not reach levels similar to the native murine adult globins (βmaj and βmin).Citation24,Citation27,Citation29 The findings suggest that multiple differences exist between the human and mouse in terms of the regulatory sequences within the human locus and the trans-acting factors responsible for regulation.
To further dissect the regulation of the human locus in the mouse, Ryan et alCitation30 used a series of human minilocus constructs consisting of the LCR linked to the Aγ- and β-globin genes, and harboring progressive deletions of the region immediately upstream of the Aγ transcriptional start site. Transgenic mice carrying deletions of upstream sequences proximal to the Aγ-globin gene showed alterations to the developmental regulation of the genes, with human β-globin appearing preferentially in the yolk sac, and loss of γ-globin transcripts in the fetal liver. Further analysis of the deleted region revealed a CACCC sequence motif in the Aγ promoter, the loss of which was responsible for the aberrant regulation of the transgenes.Citation30 Such motifs are binding sites for the KLF1 transcription factor known to be associated with the γ- to β-globin switch,Citation31 supporting the hypothesis that while high-level transcription of individual globin genes is driven by the LCR, developmental specificity is conferred by a combination of transcription factors and promoter sequence elements.
Observation of globin switching in viable cells was achieved through the creation of transgenic animals carrying fluorescent reporter constructs. Placing the coding sequence of green fluorescent protein (GFP) under the control of the human ε-globin promoter and a mini-LCR resulted in labeling of the primitive erythroblasts. Examination of the fluorescently tagged cells revealed changes in surface antigen expression as the cells matured and enucleated, demonstrating the utility of this approach for sensitive labeling of erythroid cells.Citation32
Modification of β-globin BACs to introduce fluorescent reporter genes in place of globin-coding sequences within the native genomic loci was used to produce transgenic animals with single (γ-GFP/GG mouse)Citation33 or dual (γ-GFP and β-dsRed)Citation34 reporters. GFP fluorescence traced the developmental locations of erythropoiesis (yolk sac, aorta-gonad-mesonephros, and fetal liver) in the GG mouse and progressive silencing of the reporter gene, as anticipated (). Also, in vitro drug treatment of fetal liver erythroid cells demonstrated the utility of such cells for high-throughput screening of compounds for inducers of HbF.Citation33
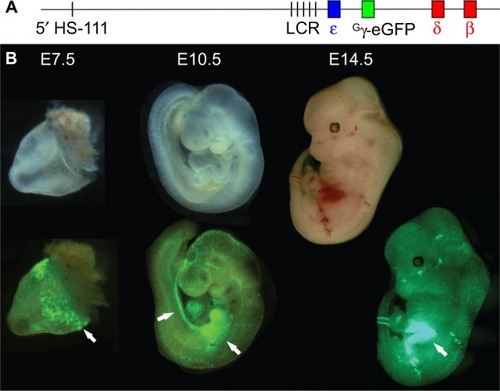
Figure 2 Visualization of sites of erythropoiesis using the Gγ-eGFP fluorescently tagged BAC reporter mouse.
Notes: (A) The human β-globin locus, carried on a BAC, was modified via recombineering to replace the Gγ and Aγ genes with that of eGFP, under the control of the Gγ promoter. (B) Transgenic mouse embryos carrying the modified β-globin locus are shown under visible light (upper) and fluorescence illumination (lower). eGFP fluorescence marks the sites yolk sac blood islands at E7.5, the aorta-gonad-mesonephros and fetal liver at E10.5, and the fetal liver at E12.5 (arrows).Citation33
Mouse deletion models of β-thalassemia
The first published model of β-thalassemia in the mouse was a spontaneously occurring mutation identified on the basis of reduced expression of the βmaj gene.Citation35 Mice homozygous for the mutation (Hbbth-1) were viable but lacked detectable βmaj protein and showed evidence of a compensatory increase in expression of the βmin gene. Hematological characteristics were reminiscent of thalassemia intermedia in the homozygotes, whereas heterozygotes showed normal blood parameters. A subsequent analysis of the Hbbth-1 mouse suggested that, in comparison to human, the decreased solubility and lack of proteolytic degradation of free mouse α-globin contributed to the severity of the Hbbth-1 phenotype.Citation36 Interestingly, a targeted disruption of the same βmaj gene (Hbbth-2) by insertion of a selectable neomycin cassette resulted in a lethal thalassemia in homozygotes at late gestation or shortly after birth.Citation37 The timing of lethality in the mice, shortly after the switch from fetal to adult β-globin expression, mirrors the postnatal appearance of symptoms in human patients as switching progresses. The authors suggest the phenotypic difference between the Hbbth-1 and Hbbth-2 mice resulted from the addition to the locus of the TK promoter within the targeting cassette. Competition for interaction with the LCR occurred between the βmaj, βmin, and TK promoters; however, only the βmin gene transcribed a functional β-globin gene. Lower levels of βmin expression were observed in Hbbth-2 than Hbbth-1, suggesting that this unintended modulation of LCR activity was responsible for the severe β-thalassemic phenotype, further supporting the one enhancer–two promoter competitive model of globin switching.
Further gene-targeting strategies lead to the creation of mice lacking both the adult βmaj and βmin genes, recapitulating the severe β0-thalassemia phenotype (Hbb0 and Hbbth-3 mice).Citation38,Citation39 As with the Hbbth-2 mutation, homozygous deletion of both adult genes was lethal at either late gestation or shortly after birth.
Significantly, no upregulation of the embryonic εy and βh1 genes was detected in the homozygous deletion animals, unlike the elevated expression of the human γ-globin genes observed in deletional HPFH. This observation indicates that the murine embryonic and adult globin genes are not in direct competition for the LCR enhancer activity. Instead it may be that the interaction with the LCR is determined by developmentally regulated transcription factors that bind specifically to either the embryonic or adult promoters, but not both. Under this hypothesis, loss of the adult genes does not result in an increased expression of the embryonic genes because the trans-acting factors responsible for positioning the adult promoters at the LCR fail to recognize the embryonic promoters.
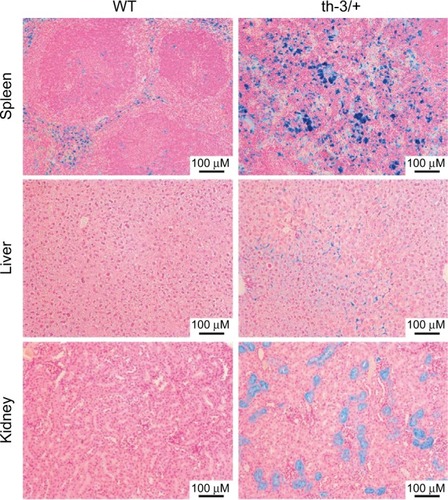
Although homozygous knockout mice were not viable, heterozygotes survived, albeit with phenotypes remarkably reminiscent of severe β-thalassemic patients, in line with the conserved structure and function of Hb between the two species. RBC Hb concentrations were reduced, cells showed microcytosis and anisopoikilocytosis, hematocrit was reduced, and reticulocytes were elevated. In addition, mice showed systemic effects similar to those observed in the clinic – splenomegaly due to extramedullary erythropoiesis, bone malformation, and iron deposition in multiple tissues including the liver (). The Hbbth-3 mice exhibit reduced expression of the iron-regulatory hormone hepcidin, a phenomenon observed in human patients also.Citation40 Repression of hepcidin stimulates duodenal iron absorption, a counter-intuitive occurrence given the iron overloaded phenotype of Hbbth-3 mice and β-thalassemia patients. This observation indicates that the Hbbth-3 mouse may be useful to investigate the more subtle defects of iron metabolism in β-thalassemia.
Figure 3 Systemic iron accumulation in the Hbbth-3/+ β-thalassemic mouse.
Abbreviation: WT, wild-type.
Of particular significance in the characterization of the Hbbth-3 mouse was the demonstration that the homozygous deletion genotype could be rescued by crossing with mice expressing high levels of human hemoglobin A (HbA).Citation38,Citation41 This confirmed the structural and functional conservation of Hb between the species, laying the groundwork for future models expressing exclusively human globin genes in conjunction with genuine SCD or β-thalassemia mutations.
Humanized murine models of β-thalassemia
Although deletion of the mouse β-globin genes resulted in phenotypes similar to the human β-thalassemias, such models lacked the human genomic sequences responsible for the condition. Therefore, therapeutic strategies intended to correctly splice mutant transcripts, or to induce expression of the γ-globins could not be tested in these animals.
The prospect of remedying splicing defects leads to the development of the Hbbth-4 mouse, incorporating a human β-globin gene integrated into the mouse locus in place of the deleted adult globin genes of Hbbth-3. The integrated human gene carried the IVS-2-654 mutation that generates an incorrectly spliced primary transcript, resulting in heterozygotes with features of β-thalassemia intermedia, while homozygous mice were not viable.Citation42 This model has been employed to demonstrate repair of the aberrant splicing in vivo and improvement of the thalassemic symptoms, by intravenous delivery of a splice-selecting oligonucleotide. The splice-selecting oligonucleotide employed was a morpholino oligomer designed to block the mutant splice site and force the correct splicing event.Citation43 When combined with α-globin knockdown to further normalize α:β globin ratios, splicing repair achieved substantial improvement in erythropoiesis.Citation44
More extensively humanized models were created through the use of bacterial recombineering techniques to introduce specific β-thalassemia mutations into human genomic fragments propagated as BACs, and the mutated loci used to generate transgenic animals. The simplicity and fidelity of recombineering permitted the generation of several genomic constructs featuring common β-thalassemia mutations (HbE, IVS1-110, and CD 41/42)Citation45–Citation49 and the creation of humanized transgenic mice incorporating mutated loci.Citation50,Citation51 When carried on a genetic background of heterozygous or homozygous deletion of the mouse adult globin genes, these models could be considered to represent a near optimal recreation of the human condition, within the limits imposed by interspecies variation. Significantly, the aberrant splicing events of the IVSI-110 and HbE mutations are recapitulated in the mouse; this finding demonstrates the functional conservation of transcript splicing between species and the suitability of the murine model to investigate methods to correct the process in humans ().Citation49,Citation51
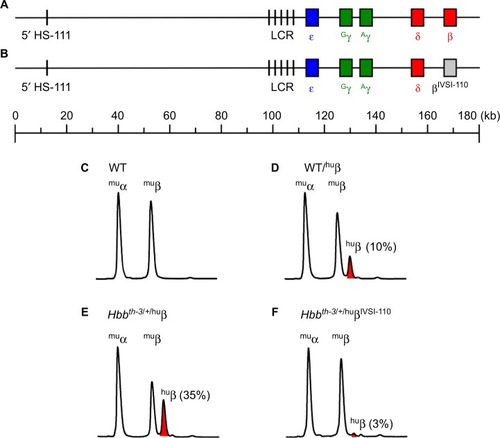
Figure 4 Recapitulation of IVSI-110 β-thalassemia splicing defect in BAC transgenic mice.
Notes: (A) BAC recombineering was used to modify the 180-kb WT human β-globin locus so as to incorporate the IVSI-110 splicing mutation within the β-globin gene (B). HPLC analysis of globin profiles from (C) WT mice showed approximately equal proportions of murine α- and β-globin (muα, muβ), whereas human β-globin made up approximately 10% of the total globins in WT mice carrying the native human β-globin locus (huβ, shown in red) (D). (E) Heterozygous Hbbth-3/+ mice carrying the WT human β-globin locus mice expressed higher levels of human β-globin, whereas (F) the presence of the IVSI-110 splicing mutation in the human β-globin locus abrogated expression substantially on the same β-thalassemic genetic background.
One further model of severe β-thalassemia was created through targeted insertion of a human Aγ-β0 (IVS1-1) cassette in place of the murine adult βmaj and βmin genes.Citation52 Unsurprisingly, heterozygotes displayed characteristics of β-thalassemia in their RBC indices as well as splenic and hepatic erythropoiesis. The authors reported an extended expression of the human Aγ gene, reminiscent of the two-stage switching in humans, and showed in a separate publication expression of the knocked-in Aγ gene in the definitive erythroid lineage.Citation53 Regardless, homozygous Aγ-β0 pups did not survive to birth, dying at ~E18.5 due to severe anemia.Citation52
Transgenic SCD models
Following the demonstration that transgenic mice expressing the human α- and β-globin genes produced functional human HbA,Citation41 humanized murine models of SCD were created. Expression of the mutated human βS-globin resulted in minimal hematological changes and little sickling in vivo, reminiscent of the heterozygous sickle cell trait and presumably due to the compensatory effect of the native mouse β-globins.Citation54,Citation55 More extensive SCD phenotypes were achieved by expression of βS variants incorporating additional mutations known to enhance the severity of the sickle phenotype (Antilles [β23Ile] and D-Punjab [β121Gln], the βSAD mouse) and crossing the human α- and βS transgenes onto a β-thalassemic background.Citation56–Citation58 Severe anemia, in vivo sickling of RBCs, and mortality in response to hypoxia was the result. Of note was the frequent occurrence of lethality in utero, due to the mid-gestational switch to expression of adult globins that occurs in the mouse. The timing of this event resulted in a fetal expression of the sickling phenotype, whereas in humans the postnatal switch from γ- to β-globin masks the presence of the sickle cell mutation until after birth.
Two further models sought to replicate SCD as extensively as possible, by generating mice carrying the genes for human α- and βS-globin on a genetic background deficient for the murine globins.Citation59,Citation60 OneCitation60 or both γ-globin genesCitation59 were incorporated into these models in an attempt to avert the gestational lethality observed previously, by taking advantage of the antisickling properties of the γ-globins and the extended expression of the human genes into the latter stages of murine fetal development. Frequent hypoxic death was observed in the hours following birth,Citation59 presumably due to severe sickle crisis induced by hypoxia in the period when newborn lungs commence functioning independently. Surviving mice reached adulthood expressing exclusively human HbS and showed a SCD phenotype closely paralleling that of human patients – in vivo sickling of RBCs in response to hypoxia, anemia with reduced hematocrit, and increased reticulocytes. The mice also exhibited the systemic symptoms of the condition, including increased weight of heart and spleen, and splenic erythropoiesis.Citation59,Citation60
Gene therapy in the mouse
As can be seen, the globin field has accumulated a panoply of mouse models intended to examine the mechanisms underlying globin switching and to recreate genetic perturbations of globin synthesis (summarized in ). As with all models, the value of the mouse lies in the degree to which it recapitulates the functioning of human biology. The structural and functional conservation of the β-globins between mice and humans is demonstrated by the formation of functional human Hb in transgenic mice and the recreation of the symptoms of the β-hemoglobinopathies in mice carrying SCD and β-thalassemia genotypes. In addition, human β- and mouse α-globin proteins can combine to form functional chimeric Hb. This functional conservation is essential for testing gene therapy vectors expressing the human protein in mouse models of the β-hemoglobinopathies.
Table 1 Summary of murine models of β-thalassemia and sickle cell disease
Early gene therapy research examined expression of the human β-globin gene when driven by minimal LCR constructs in order to determine what sequence elements are essential to drive high-level transgene expression.Citation61 Similar work has been undertaken to determine the levels of γ-globin expression that are sufficient for therapeutic effects.Citation62 A genuine strategy for gene therapy of the β-hemoglobinopathies was first successfully demonstrated in the Hbbth-3/+ mouse, using a lentivirus for ex vivo delivery of a functional β-globin minigene into bone marrow hematopoietic stem cells (HSCs) followed by transplantation of the modified cells into Hbbth-3/+ recipient animals.Citation63 Upon reconstitution of the hematopoietic system in vivo, Hbbth-3/+ stem cells carrying the lentiviral vector (LV) provided a substantial improvement in multiple symptoms – increased hematocrit and Hb concentrations – while reticulocytes and RBC morphological abnormalities associated with the thalassemia phenotype were reduced. Long-term studies of the LV-treated animals showed sustained improvement in Hb levels and reduced hepatic iron accumulation.Citation64 The same study compared the expression of the β-globin transgene in vectors featuring minimal or more extensive LCR sequences; unsurprisingly, larger LCR fragments were more effective drivers of expression.
A separately developed LV achieved extensive rescue of the SCD phenotype in βSAD and BERK mice through the expression of a variant β-globin created to inhibit the Hb polymerization of the condition.Citation65 Significant reduction of RBC sickling was observed, along with improvements in hematological parameters. The authors noted the necessity for high viral titers to achieve the pancellular expression of the transgene necessary to fully ameliorate the SCD phenotype.
Further gene transfer studies confirmed the utility of LVs for the delivery of β-globin transgenes into HSCs and the subsequent improvement in symptoms in SCD and β-thalassemic mice following engraftment of transduced cells.Citation66,Citation67 Significantly, long-term analysis of mice following gene transfer therapy confirmed the capacity of LVs to maintain expression of β- or γ-globin transgenes, an essential prerequisite for clinical success.Citation64,Citation68,Citation69 Such vectors have now passed into human trials, where they have achieved relief of acute and chronic symptoms of β-thalassemia and long-term transfusion independence.Citation70,Citation71 Further trials in β-thalassemia and SCD patients are ongoing.Citation72
Moving beyond classical viral gene replacement therapy, the development of induced pluripotent stem cells raised the possibility of repairing the genetic defect in cells derived from individual patients. The repaired cells might then be differentiated in vitro into HSCs and returned to the patient where they will give rise to RBCs expressing the repaired β-globin gene. Such an approach was shown to be possible in a SCD mouse, albeit in the short term as the field has yet to achieve the generation of truly long-term repopulating HSCs from induced pluripotent stem cells.Citation73
Animal models to manipulate globin switching
Examination of β-globin gene switching in the mouse has provided tremendous advances in our understanding of gene regulation, a primary driver of early investigations of the locus. The fundamental processes at work in widely divergent species are sufficiently similar to ensure that study of the mouse, and even the chicken, has been a source of enormous insight into fundamental human biology. Such work continues to date, with current interest focusing upon chromatin and epigenetic regulation as components of the switching process.
The promise of gene therapy to provide a cure for the β-hemoglobinopathies now seems close to becoming a reality, at least technically. At present, however, gene therapy fails to deliver universal treatment for nontechnical reasons, specifically those relating to economics. Clinical gene therapy is a technologically demanding field, restricted to a small number of centers worldwide. The costs associated with stem cell transfer are substantial, with single treatments costing in excess of 100,000–200,000 USD,Citation74 while the first gene therapy treatment in the western world for lipoprotein lipase deficiency costs around $1 million per treatment.Citation75 These facts collide with the reality of the worldwide distribution of SCD and β-thalassemia patients. World Health Organization figures demonstrate that the global burden of births affected by these conditions is carried primarily by African populations, located in regions where annual per capita expenditure on health is one to two orders of magnitude lower than in the most developed nations.Citation76 This economic reality therefore demands cheaper therapies, driving the field to search for small molecules capable of reactivating γ-globin expression and suitable for long-term administration. At present, hydroxyurea is the only agent approved for reactivation of γ-globin expression in SCD and β-thalassemia; however, responses are variable, and there are no known biomarkers to definitively predict response.Citation77
The use of mouse models to search for strategies and compounds to reactivate γ-globin expression is predicated upon the assumption that the regulatory mechanisms responsible are substantially conserved between the species. The flaws in this assumption relate in this case to the single developmental β-globin switch in rodents and the lack of a fetal stage Hb (HbF). This difference is most clearly apparent in BAC and YAC transgenic mice, where the native arrangement of large genomic transgenes permits researchers to discount the possibility of artifacts resulting from the use of minimal constructs with reorganized or absent regulatory elements.
In humans, the expression of γ-globin commences with the onset of definitive erythropoiesis, in concert with silencing of the ε-globin gene. β-Globin BAC and YAC transgenic mice reveal the evolutionary relationship between murine βh1 and human γ-globin genes, in that the two are expressed in parallel in the mouse, commencing in the primitive erythroid precursors of the yolk sac.Citation23,Citation25 However, while βh1-globin expression is silenced by murine mid-gestation, when definitive erythropoiesis is occurring in the fetal liver, transgenic mice exhibit substantial human γ-globin transcription in erythroid tissue at this time point. The earlier onset of γ-globin expression and the delay in silencing relative to εy and βh1-globin have been observed in multiple independently generated models, confirming the biological divergence between the two species ().
Examination of this phenomenon further illustrated the regulation of the human embryonic γ-globin gene as a fetal/embryonic gene in the mouse. Separation of primitive and definitive erythroid cells from E13.5 mice carrying a human β-globin YAC transgene demonstrated substantial elevation of γ-globin expression in the primitive cells relative to the definitive.Citation78 Microscopic examination of fetal livers revealed the presence of nucleated primitive erythroblasts expressing γ-globin at E13.5, whereas expression was largely absent in enucleated definitive cells at the same time point. Interestingly, a small proportion of adult RBCs contain transgenic γ-globin protein, similar to the infrequent F-cells detected in adult humans and demonstrating that the γ-globin gene is also expressed to some degree in murine definitive erythroid tissue.Citation23 Expression of γ-globin was also observed in the definitive erythroid lineage of the Aγ-β knock-in mouse.Citation53
These interspecies differences present a difficulty to researchers seeking to use the mouse as a model to dissect the regulation of γ-globin and devise new therapies targeting the process. Encouragingly, reports abound in the literature describing regulators of γ-globin that demonstrate conserved function in both mouse models of globin switching and primary human erythroid cells.Citation31,Citation79–Citation82 Such findings demonstrate the conservation of many aspects of globin switching between human and mouse, and, accordingly, significant efforts have been put in place to devise reporter animals and to conduct chemical screens for novel inducers of HbF in mouse cells.Citation33,Citation34,Citation83
To move beyond the mouse, some researchers have made use of the baboon to study globin switching in primates. Although the baboon closely reiterates the structure and switching of the human β-globin locus, the inherent complexities of primate research have limited the number of such studies. Nevertheless, valuable insights have been gained, particularly in preclinical testing of novel therapeutic inducers.Citation84–Citation86
Conclusion
The development and availability of biological resources and sophisticated genomic technologies to manipulate the mouse genome has enhanced our understanding of the regulatory network responsible for globin expression. Importantly, a better understanding of these developmental events may allow the identification of key targets for therapeutic intervention. The mouse has been shown to be a physiologically relevant mammalian system, which recreates specific disease phenotypes associated with β-hemoglobinopathies. Given the complex pathophysiology of β-hemoglobinopathies and our current incomplete understanding of globin gene regulation in humans, the mouse model will continue to provide valuable insight into the genetic basis of human disease. Despite these decades of work and the elucidation of the molecular basis of SCD and β-thalassemia, improvement in the clinical management of these conditions has been slow. The challenge now is to move beyond the creation of model systems and to identify novel pharmacological and genetic approaches that are suitable for widespread application to the multitude of patients worldwide who are living with the β-hemoglobinopathies.
Note concerning gene nomenclature. The authors have used non-standard nomenclature for gene names, reflecting that which is in common usage amongst researchers in the globin field. For clarity the standardised gene names are as follows: Human β-globin, HBB; δ, HBD; Aγ, HBG1; Gγ, HBG2; ε, HBE; ζ, HBZ; α, HBA1 & HBA2. Murine βmaj-globin, Hbb-b1; βmin, Hbb-b2; βh1, Hbb-bh1; εy, Hbb-y.
Acknowledgments
This work was supported by the Australian National Health and Medical Research Council, the Murdoch Childrens Research Institute, the Victorian Government’s Operational Infrastructure Support Program, and Thalassaemia Australia.
Disclosure
The authors report no conflicts of interest in this work.
References
- OrkinSHZonLIHematopoiesis: an evolving paradigm for stem cell biologyCell2008132463164418295580
- BaronMHIsernJFraserSTThe embryonic origins of erythropoiesis in mammalsBlood2012119214828483722337720
- WeatherallDJThe inherited diseases of hemoglobin are an emerging global health burdenBlood2010115224331433620233970
- IngramVMA specific chemical difference between the globins of normal human and sickle-cell anaemia haemoglobinNature1956178453779279413369537
- PaulingLItanoHASickle cell anemia a molecular diseaseScience1949110286554354815395398
- IngramVMGene mutations in human haemoglobin: the chemical difference between normal and sickle cell haemoglobinNature1957180458132632813464827
- HalbrechtIKlibanskiCIdentification of a new normal embryonic haemoglobinNature1956178453779479513369538
- WatsonJThe significance of the paucity of sickle cells in newborn Negro infantsAm J Med Sci1948215441942318107723
- FessasPStamatoyannopoulosGHereditary persistence of fetal hemoglobin in Greece: a study and a comparisonBlood196424322324014214133
- KulozikAEYarwoodNJonesRWThe Corfu delta beta zero thalassemia: a small deletion acts at a distance to selectively abolish beta globin gene expressionBlood19887124574622827815
- SankaranVGXuJOrkinSHAdvances in the understanding of haemoglobin switchingBr J Haematol2010149218119420201948
- ChadaKMagramJRaphaelKRadiceGLacyECostantiniFSpecific expression of a foreign beta-globin gene in erythroid cells of transgenic miceNature198531460093773803982505
- MagramJChadaKCostantiniFDevelopmental regulation of a cloned adult beta-globin gene in transgenic miceNature198531560173383403858676
- ChadaKMagramJCostantiniFAn embryonic pattern of expression of a human fetal globin gene in transgenic miceNature1986319605568568910.1038/319685a03951540
- GrosveldFvan AssendelftGBGreavesDRKolliasGPosition-independent, high-level expression of the human beta-globin gene in transgenic miceCell19875169759853690667
- EnverTRaichNEbensAJPapayannopoulouTCostantiniFStamatoyannopoulosGDevelopmental regulation of human fetal-to-adult globin gene switching in transgenic miceNature199034462643093132314472
- StamatoyannopoulosGJosephsonBZhangJWLiQDevelopmental regulation of human gamma-globin genes in transgenic miceMol Cell Biol19931312763676448246980
- BehringerRRRyanTMPalmiterRDBrinsterRLTownesTMHuman gamma- to beta-globin gene switching in transgenic miceGenes Dev1990433803891692558
- ChoiOREngelJDDevelopmental regulation of beta-globin gene switchingCell198855117263167976
- NickolJMFelsenfeldGBidirectional control of the chicken beta- and epsilon-globin genes by a shared enhancerProc Natl Acad Sci U S A1988858254825523357880
- ForgetBGMolecular basis of hereditary persistence of fetal hemoglobinAnn N Y Acad Sci199885038449668525
- BankAO’NeillDLopezRRole of intergenic human gamma-delta-globin sequences in human hemoglobin switching and reactivation of fetal hemoglobin in adult erythroid cellsAnn N Y Acad Sci20051054485416339651
- PetersonKRCleggCHHuxleyCTransgenic mice containing a 248-kb yeast artificial chromosome carrying the human beta-globin locus display proper developmental control of human globin genesProc Natl Acad Sci U S A19939016759375978356061
- PorcuSKitamuraMWitkowskaEThe human beta globin locus introduced by YAC transfer exhibits a specific and reproducible pattern of developmental regulation in transgenic miceBlood19979011460246099373272
- StrouboulisJDillonNGrosveldFDevelopmental regulation of a complete 70-kb human beta-globin locus in transgenic miceGenes Dev1992610185718641383089
- GaenslerKMKitamuraMKanYWGerm-line transmission and developmental regulation of a 150-kb yeast artificial chromosome containing the human beta-globin locus in transgenic miceProc Natl Acad Sci U S A1993902311381113858248258
- KaufmanRMPhamCTLeyTJTransgenic analysis of a 100-kb human beta-globin cluster-containing DNA fragment propagated as a bacterial artificial chromosomeBlood19999493178318410556205
- PetersonKRLiQLCleggCHUse of yeast artificial chromosomes (YACs) in studies of mammalian development: production of beta-globin locus YAC mice carrying human globin developmental mutantsProc Natl Acad Sci U S A19959212565556597539923
- VadolasJWardanHBosmansMTransgene copy number-dependent rescue of murine beta-globin knockout mice carrying a 183 kb human beta-globin BAC genomic fragmentBiochim Biophys Acta20051728315016215820143
- RyanTMSunCWRenJTownesTMHuman gamma-globin gene promoter element regulates human beta-globin gene developmental specificityNucleic Acids Res200028142736274010908330
- TallackMRPerkinsACThree fingers on the switch: Krüppel-like factor 1 regulation of γ-globin to β-globin gene switchingCurr Opin Hematol201320319320023474875
- FraserSTIsernJBaronMHMaturation and enucleation of primitive erythroblasts during mouse embryogenesis is accompanied by changes in cell-surface antigen expressionBlood2007109134335216940424
- McCollBKaoBRLourthaiPAn in vivo model for analysis of developmental erythropoiesis and globin gene regulationFASEB J Off Publ Fed Am Soc Exp Biol201428523062317
- PapadopoulosPGutiérrezLvan der LindenRA dual reporter mouse model of the human β-globin locus: applications and limitationsPloS One2012712e5127223272095
- SkowLCBurkhartBAJohnsonFMA mouse model for beta-thalassemiaCell1983343104310526313205
- Rouyer-FessardPLeroy-ViardKDomengetCMradABeuzardYMouse beta thalassemia, a model for the membrane defects of erythrocytes in the human diseaseJ Biol Chem19902653320247202512243088
- SheheeWROliverPSmithiesOLethal thalassemia after insertional disruption of the mouse major adult beta-globin geneProc Natl Acad Sci U S A1993908317731818475058
- CiavattaDJRyanTMFarmerSCTownesTMMouse model of human beta zero thalassemia: targeted deletion of the mouse beta maj- and beta min-globin genes in embryonic stem cellsProc Natl Acad Sci U S A19959220925992637568113
- YangBKirbySLewisJDetloffPJMaedaNSmithiesOA mouse model for beta 0-thalassemiaProc Natl Acad Sci U S A1995922511608116128524813
- Weizer-SternOAdamskyKAmariglioNmRNA expression of iron regulatory genes in beta-thalassemia intermedia and beta-thalassemia major mouse modelsAm J Hematol200681747948316755567
- BehringerRRRyanTMReillyMPSynthesis of functional human hemoglobin in transgenic miceScience198924549219719732772649
- LewisJYangBKimRA common human beta globin splicing mutation modeled in miceBlood1998916215221569490703
- SvastiSSuwanmaneeTFucharoenSRNA repair restores hemoglobin expression in IVS2-654 thalassemic miceProc Natl Acad Sci U S A200910641205121019164558
- XieSYLiWRenZRHuangSZZengFZengYTCorrection of β654-thalassaemia mice using direct intravenous injection of siRNA and antisense RNA vectorsInt J Hematol201193330131021369857
- JamsaiDNefedovMNarayananKInsertion of common mutations into the human beta-globin locus using GET Recombination and an EcoRI endonuclease counterselection cassetteJ Biotechnol200310111912523964
- NefedovMWilliamsonRIoannouPAInsertion of disease-causing mutations in BACs by homologous recombination in Escherichia coliNucleic Acids Res20002817E7910954612
- JamsaiDOrfordMNefedovMFucharoenSWilliamsonRIoannouPATargeted modification of a human beta-globin locus BAC clone using GET recombination and an I-Scei counterselection cassetteGenomics2003821687712809677
- JamsaiDOrfordMFucharoenSWilliamsonRIoannouPAInsertion of modifications in the beta-globin locus using GET recombination with single-stranded oligonucleotides and denatured PCR fragmentsMol Biotechnol2003231293612611267
- JamsaiDZaibakFVadolasJA humanized BAC transgenic/knockout mouse model for HbE/beta-thalassemiaGenomics200688330931516631345
- JamsaiDZaibakFKhongniumWA humanized mouse model for a common beta0-thalassemia mutationGenomics200585445346115780748
- VadolasJNefedovMWardanHHumanized beta-thalassemia mouse model containing the common IVSI-110 splicing mutationJ Biol Chem2006281117399740516421096
- HuoYMcConnellSCLiuSRHumanized mouse model of cooley’s anemiaJ Biol Chem200928484889489619098001
- McConnellSCHuoYLiuSRyanTMHuman globin knock-in mice complete fetal-to-adult hemoglobin switching in postnatal developmentMol Cell Biol201131487688321173165
- RyanTMTownesTMReillyMPHuman sickle hemoglobin in transgenic miceScience199024749425665682154033
- GreavesDRFraserPVidalMAA transgenic mouse model of sickle cell disorderNature199034362541831852296310
- RubinEMWitkowskaHESpanglerEHypoxia-induced in vivo sickling of transgenic mouse red cellsJ Clin Invest19918726396471991848
- TrudelMSaadaneNGarelMCTowards a transgenic mouse model of sickle cell disease: hemoglobin SADEMBO J19911011315731651915288
- FabryMESenguptaASuzukaSMA second generation transgenic mouse model expressing both hemoglobin S (HbS) and HbS-Antilles results in increased phenotypic severityBlood1995866241924287662990
- PásztyCBrionCMManciETransgenic knockout mice with exclusively human sickle hemoglobin and sickle cell diseaseScience199727853398768789346488
- RyanTMCiavattaDJTownesTMKnockout-transgenic mouse model of sickle cell diseaseScience199727853398738769346487
- EllisJPasceriPTan-UnKCEvaluation of beta-globin gene therapy constructs in single copy transgenic miceNucleic Acids Res1997256129613029092642
- NishinoTCaoHStamatoyannopoulosGEmeryDWEffects of human gamma-globin in murine beta-thalassaemiaBr J Haematol2006134110010816803575
- MayCRivellaSCallegariJTherapeutic haemoglobin synthesis in beta-thalassaemic mice expressing lentivirus-encoded human beta-globinNature20004066791828610894546
- MayCRivellaSChadburnASadelainMSuccessful treatment of murine beta-thalassemia intermedia by transfer of the human beta-globin geneBlood20029961902190811877258
- PawliukRWestermanKAFabryMECorrection of sickle cell disease in transgenic mouse models by gene therapyScience200129455502368237111743206
- PuthenveetilGScholesJCarbonellDSuccessful correction of the human beta-thalassemia major phenotype using a lentiviral vectorBlood2004104123445345315292064
- PerumbetiAHigashimotoTUrbinatiFA novel human gamma-globin gene vector for genetic correction of sickle cell anemia in a humanized sickle mouse model: critical determinants for successful correctionBlood200911461174118519474450
- MiccioACesariRLottiFIn vivo selection of genetically modified erythroblastic progenitors leads to long-term correction of beta-thalassemiaProc Natl Acad Sci U S A200810530105471055218650378
- PestinaTIHargrovePWJayDGrayJTBoydKMPersonsDACorrection of murine sickle cell disease using gamma-globin lentiviral vectors to mediate high-level expression of fetal hemoglobinMol Ther J Am Soc Gene Ther2009172245252
- Cavazzana-CalvoMPayenENegreOTransfusion independence and HMGA2 activation after gene therapy of human β-thalassaemiaNature2010467731331832220844535
- NegreOBartholomaeCBeuzardYPreclinical evaluation of efficacy and safety of an improved lentiviral vector for the treatment of β-thalassemia and sickle cell diseaseCurr Gene Ther2015151648125429463
- NegreOEggimannAVBeuzardYGene therapy of the β-Hemoglobinopathies by lentiviral transfer of the β(A(T87Q))-Globin geneHum Gene Ther201627214816526886832
- HannaJWernigMMarkoulakiSTreatment of sickle cell anemia mouse model with iPS cells generated from autologous skinScience200731858581920192318063756
- KheraNZeliadtSBLeeSJEconomics of hematopoietic cell transplantationBlood201212081545155122700725
- Ylä-HerttualaSGlybera’s second act: the curtain rises on the high cost of therapyMol Ther201523221721825633169
- World Health OrganizationGlobal Epidemiology of Haemoglobin Disorders and Derived Service IndicatorsWorld Health Organization2008 Available from: http://www.who.int/bulletin/volumes/86/6/06-036673-ab/en/
- ZimmermanSASchultzWHDavisJSSustained long-term hematologic efficacy of hydroxyurea at maximum tolerated dose in children with sickle cell diseaseBlood200410362039204514630791
- SankaranVGXuJRagoczyTDevelopmental and species-divergent globin switching are driven by BCL11ANature200946072591093109719657335
- XuJPengCSankaranVGCorrection of sickle cell disease in adult mice by interference with fetal hemoglobin silencingScience2011334605899399621998251
- ShiLCuiSEngelJDTanabeOLysine-specific demethylase 1 is a therapeutic target for fetal hemoglobin inductionNat Med201319329129423416702
- XuJBauerDEKerenyiMACorepressor-dependent silencing of fetal hemoglobin expression by BCL11AProc Natl Acad Sci U S A2013110166518652323576758
- AmayaMDesaiMGnanapragasamMNMi2β-mediated silencing of the fetal γ-globin gene in adult erythroid cellsBlood2013121173493350123444401
- PetersonKRCostaFCFedosyukHA cell-based high-throughput screen for novel chemical inducers of fetal hemoglobin for treatment of hemoglobinopathiesPloS One201499e10700625225870
- BoosalisMSCastanedaSATrudelMNovel therapeutic candidates, identified by molecular modeling, induce γ-globin gene expression in vivoBlood Cells Mol Dis201147210711621641240
- BoosalisMSSangermanJIWhiteGLNovel inducers of fetal globin identified through High Throughput Screening (HTS) are active in vivo in anemic baboons and transgenic micePloS One20151012e014466026713848
- RiversAVaitkusKRuizMARN-1, a potent and selective lysine-specific demethylase 1 inhibitor, increases γ-globin expression, F reticulocytes, and F cells in a sickle cell disease mouse modelExp Hematol201543754655325931013
- TrudelMDe PaepeMEChrétienNSickle cell disease of transgenic SAD miceBlood1994849318931977949191