Abstract
The most common subtypes of primary cutaneous T-cell lymphoma (CTCL) are mycosis fungoides and Sézary syndrome. Clinical manifestations and prognosis in CTCL are highly variable. Improving the management of this incurable disease with limited toxicity is an active area of research. Romidepsin is a novel, well-tolerated histone deacetylase inhibitor with promising activity against advanced stages of CTCL. In November 2009, it was approved by the US Food and Drug Administration for the treatment of CTCL in patients who have received at least one prior systemic therapy. This review focuses on the activity, pharmacology, and safety of romidepsin for the treatment of CTCL.
Keywords:
Introduction
Cutaneous T-cell lymphoma (CTCL) is a rare group of mature T-cell lymphomas primarily presenting in the skin. Between 2000 and 3000 new cases of CTCL occur in the US each year, with mycosis fungoides (MF) being the predominant subtype.Citation1 Skin manifestations of MF include patches, plaques, tumors, and erythroderma. MF is categorized as limited stage by plaques or patches limited to the skin (IA, IB, and IIA) and advanced stage by cutaneous tumors and involvement of the blood, lymph nodes, bone marrow, or visceral organs (IIB to IVB).Citation2 Sézary syndrome (SS), the leukemic variant of MF, is characterized by generalized erythroderma and abnormal lymphoid cells in the blood.Citation3 Patients with limited stage disease frequently have an indolent clinical course and may be effectively treated with skin-directed therapies, including topical nitrogen mustard or psoralen plus ultraviolet A therapy.Citation4 Treatment of advanced stage disease includes systemic therapy with α interferon, bexarotene, photopheresis, denileukin diftitox, vorinostat, cytotoxic chemotherapy, or combined modality therapies. However, in most patients, responses are seldom durable. This has led to a need to develop new therapies with targeted mechanisms of action and acceptable safety profiles.
Romidepsin (ISTODAX® [Astellas Pharma Inc., Tokyo, Japan], depsipeptide, FR901228, FK228) is a promising new agent that has shown remarkable activity in the treatment of T-cell lymphomas in preclinical studies and early-phase clinical trials. In November 2009, it was granted approval by the US Food and Drug Administration (FDA) for the treatment of CTCL in patients who have received at least one prior systemic therapy. It belongs to the new class of antineoplastic agents known as histone deacetylase inhibitors (HDACis) that are known to affect gene and protein function by epigenetic modulation. Vorinostat, another HDACi, is approved in the US for the treatment of CTCL in patients who have failed at least two prior systemic therapies. HDACis have shown remarkable signal of activity in some T-cell malignancies such as CTCL and peripheral T-cell lymphoma, and several of these agents, including belinostat, panobinostat, and entinostat, are in various stages of clinical trials. The results are summarized in and .
Table 1 Summary of the histone deacetylase (HDAC) inhibitors tested in cutaneous T-cell lymphoma
Table 2 Outcomes of histone deacetylase inhibitors in CTCL
Discovery, structure, and mechanism of action
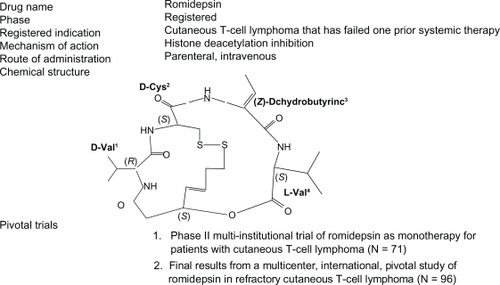
Romidepsin was isolated from the fermentation broth of a Gram-negative, nonsporing coccobacillus Chromabacterium violaceum strain WB968 by Fujisawa Pharmaceutical Company in Japan in an effort to identify compounds that can selectively reverse the phenotype of the ras transformed cells.Citation5,Citation6 Romidepsin was found to have the ability to reverse the morphology of the Ha-ras transformed cells to a normal phenotype. Hence, it was isolated and purified and was found to be a colorless prism with the structure of (E)-(1S, 4S, 10S, 21R)-7-[(Z)-ethylidene]-4, 21-diisopropyl-2-oxa-12, 13-dithia-5, 8, 20, 23-tetraazabicyclo [8, 7, 6]-tricos-16-ene 3,6,9,19,22-pentanone, as shown in . It was lipophilic and possessed a unique noncystine disulfide linkage. Nakajima et al showed that romidepsin caused the arrest of the cell cycle at both G1 and G2/M phases and the induction of internucleosomal breakdown of chromatin characteristic of apoptosis.Citation7 They also demonstrated that it inhibited intracellular histone deacetylase activity, leading to marked accumulation of acetylated histone (H1, H2A, H2B, H3, and H4) in the cell, and established its role as a novel HDACi. Furumai et al demonstrated that romidepsin circulates in the serum in its inactive form and penetrates the cell membrane effectively given its hydrophobic nature.Citation8 However, on entering the tumor cell, it gets converted to an active, reduced form (redFK) by intracellular antioxidants involving glutathione. It has been proposed that one of the sulfhydryl groups of the reduced form interacts with the zinc ions in the active site of the class I HDAC enzymes, leading to potent inhibition of enzymatic activity of both class I and II HDACs in vivo, but its activity against HDAC4 and HDAC6 class II enzymes remains weak. The prodrug form of romidepsin has a half-life of >12 hours, whereas the redFK form has a half-life of 0.54 hours. This clearly demonstrates that romidepsin is quite stable in the prodrug form in blood, which may be responsible for its strong in vivo HDAC inhibition.
Figure 1 Structure of romidepsin.
It is now well established that modulation of histone and protein acetylation alters pathways that promote proliferation, angiogenesis, and survival in cancer cells. A common finding in malignant cells is the high level of expression of HDAC isoenzymes and a corresponding hypoacetylation of histones. HDACis work through myriad different mechanisms, including i) upregulation of cell-dependent kinase (cdk) inhibitors like p21/p27 and downregulation of cyclinD1; ii) acetylation of nonhistone proteins, including STAT-3, RelA/p65, p53, HIF-1α, Bcl-6, and Hsp 90, in a way that modulates their activity and thus their ability to regulate cell growth and survival; iii) direct activation of apoptotic pathways by affecting the balance between the antiapoptotic proteins like Bcl-2 and the proapapototic proteins like Bax and Bak; iv) enhanced production of reactive oxygen species along with increased thioredosin levels; v) enhanced antitumor immunity through enhancement of TRAIL or upregulation of antigen expression, which could facilitate cancer cell recognition; and vi) disruption of DNA repair through acetylation or downregulation of proteins such as Ku70, Ku86, BRCA1, and RAD51.Citation9,Citation10 Despite these pleiotropic effects, it has been difficult to assign a precise mechanistic basis to their anticancer effects in any particular tumor type, let alone CTCL. It is peculiar that romidepsin appears to have a class effect in CTCL in contrast to other subtypes of lymphoproliferative neoplasms. Gene expression profiling on paired tissue samples and studies of select biomarkers, including gene activation with HDACis, has shown that up to 5%–10% of the genome can be affected by HDACis. In one study, the genes that were consistently affected included genes affecting cell cycle (CCNDI, IGFI), apoptosis (septin10, TEF, SORBBS2), angiogenesis (GUCY1A1, ANGPT1), and immune modulation (LAIR1).Citation11
Preclinical activity
Romidepsin demonstrated potent cytotoxicity against human tumor cell lines and in vivo efficacy against both human tumor xenografts and murine tumors.Citation5,Citation6 Many human tumors appear to be associated with the expression of activated ras genes. Romidepsin was found to reverse the morphology of the Ha-ras transfected mouse NIH/3T3 (Ras1) cells to that of the parent, thereby suggesting inhibitory effects on the growth of tumor cells and potential as an anticancer agent.Citation5 This was through a decrease in the mRNA expression of the c-myc oncogene in Ras1 cells, but it had no effect on H-ras mRNA expression, implying various other potential mechanisms such as activation of the Raf/MEK/ERK axis or the PI3K/Akt/GSK-3 pathway, which needs validation in preclinical and correlative clinical studies. In vitro, it induced cell cycle arrest in both G1 and G2/M phases and apoptosis in a number of human tumor cell lines such as A549, PC-9 lung adenocarcinoma, PC-1 and PC-10 lung squamous cell carcinoma, ADH, LX-1 small cell lung carcinoma, MKN28, MKN74 stomach adenocarcinoma, MCF-7, ZR-75–1 mammary adenocarcinoma, Colo201, and SW480 colon adenocarcinoma at low ng/mL concentrations.Citation6 Romidepsin exhibited weak cytoxicity against human normal endothelial cell HE-9, mouse fibroblast Balb/c3T3, and NIH3T3 and demonstrated no activity against human and mouse normal fibroblast cells, thereby suggesting selectivity. Other in vitro studies have revealed antiproliferative effects of romidepsin against B-cell chronic lymphocytic leukemia (CLL) cell lines,Citation12 T-cell lymphoma cells,Citation13 and multiple myeloma cells.Citation14 In vivo, it was found to prolong the life of mice bearing murine ascitic tumors, murine leukemias, and melanoma and inhibited the growth of murine solid tumors such as colon cancer, sarcomas, and human solid tumors implanted in normal and nude mice.Citation6 When given intravenously every 4 days, it inhibited the growth of several human tumor xenografts such as MX-1 breast carcinoma, LC-6 lung carcinoma, LU-65 large cell lung carcinoma, and SC-6 stomach adenocarcinoma.Citation6 This antitumor activity was confirmed by the National Cancer Institute (NCI), and the agent was assigned the NSC number NSC-630176 for further clinical development.
Romidepsin showed a lack of cross-resistance to several commonly used cytotoxic agents such as vincristine, 5-fluorouracil, mitomycin C, and cyclophosphamide and was identified as a P-glycoprotein (Pgp) and multidrug resistance-associated protein (MRP-1) substrate by Xiao et al.Citation15 It is well established that Pgp and MRP-1 are associated with resistance to anticancer drugs. With the help of a romidepsin-resistant cell line (HCT15R) developed from HCT15 colon carcinoma, it was confirmed that efflux of romidepsin via Pgp induction is the major mechanism for acquired romidepsin resistance. These data need to be considered when romidepsin is used to pretreat patients who are receiving Pgp substrate drugs. This property also raises the possibility of combining romidepsin with Pgp inhibitors in developing treatment strategies for patients with CTCL or using Pgp inhibitors to reverse the resistance to romidepsin. As a substrate for Pgp-mediated drug efflux, romidepsin would likely not penetrate into the central nervous system, as opposed to the other HDACis, which are not Pgp substrates.Citation16,Citation17
Johnstone et al demonstrated that romidepsin induced the accumulation of 4n DNA content, a G2 cell cycle checkpoint in the acute T-cell leukemia cell line CEM-CCRF, and caused apoptosis via activation of the intrinsic apoptotic pathway, which was accompanied by the cleavage and activation of caspase-2 and Bid.Citation18 Romidepsin-induced mitochondrial damage and apoptosis were inhibited by overexpression of Bcl-2 but not by the polycaspase inhibitor (zVAD-fmk). Moreover, induction of a G1-S checkpoint through overexpression of p16INK4A or suppression of de novo protein synthesis inhibited romidepsin-induced cell death. While romidepsin induced the activation of the proapoptotic Bcl-2 protein, Bid cleavage in response to romidepsin was significantly attenuated by zVAD-fmk. Bid lies upstream of mitochondrial membrane disruption and is an important proapoptotic molecule. This effect has not been demonstrated with other HDACis such as oxamflatin or SAHA (vorinostat), demonstrating the ability of these drugs to induce apoptosis in the absence of activation of certain caspases and highlighting functional differences between these structurally dissimilar compounds that are thought to have similar intracellular targets. It was also found that romidepsin was a substrate of Pgp and hence less effective against Pgp-expressing tumor cells as opposed to oxamflatin and SAHA. Subsequently, Johnstone et al used a Eμ-myc mouse model of B-cell lymphoma and showed that romidepsin had the ability to kill Eμ-myc lymphomas that overexpress Bcl-2 with delayed kinetics of cell death and could mediate therapeutic responses against these lymphomas.Citation19 Of interest, they noted that Eμ-myc cells that overexpressed Bcl-XL were resistant to romidepsin as opposed to SAHA, thereby supporting the hypothesis that HDACis may have subtle yet potentially important differences in molecular activities.
Pharmacokinetics and pharmacodynamics of romidepsin
The current infusion schedule of administering romidepsin over 4 hours was based on the safety and efficacy of the 4-hour intravenous infusion over the 24-hour infusion observed in Beagle dogs and rats, following which this infusion time was incorporated for further studies in Phase I clinical trials.Citation20,Citation21 Two Phase I trials were carried out simultaneously to determine the maximum tolerated dose (MTD) and toxicity profile of the 4-hour infusion of romidepsin and to characterize its pharmacokinetics. In the first trial performed by investigators at the NCI, the agent was given intermittently on a day 1 and day 5 schedule every 21 days.Citation22 Thirty-seven patients with advanced or primary refractory neoplasms were administered the drug at that schedule. Based on the preclinical toxicology data, the starting dose of the trial was chosen as 1 mg/m2, and dose escalations proceeded through a total of eight dose levels to a maximum of 24.9 mg/m2. The MTD was defined at 17.8 mg/m2. Biologically active serum concentrations were achieved with this dose, and one partial response was observed. Dose-limiting toxicities (DLTs) included grade 3 fatigue (three patients), grade 3 nausea and vomiting (one patient), grade 4 thrombocytopenia (two patients), and grade 4 cardiac arrhythmia (one patient, atrial fibrillation). The mean volume of distribution, clearance, distribution half-life (t1/2α), and elimination half-life (t1/2β) at a dose of 17.8 mg/m2 were 8.6 L/m2, 11.6 L/h/m2, 0.42 hours, and 8.1 hours, respectively. The mean maximum plasma concentration (Cmax) at the MTD dose was 472.6 ng/mL. Furthermore, romidepsin pharmacokinetics was not significantly different when comparing the first cycle of a dose level with a subsequent cycle of the same dose. As expected, an increased dose led to increased Cmax and area under the curve (AUC). Pharmacokinetics was not affected by repeated dosing. In general, romidepsin was well tolerated. The significant cardiac toxicity seen in preclinical studies was not observed, and the dose suggested for Phase II was 17.8 mg/m2 administered on day 1 and day 5 of a 21-day cycle. The other Phase I trial conducted at the NCI utilized a different schedule in which romidepsin was administered as a 4-hour infusion given weekly for 3 weeks followed by 1 week of rest in 33 patients with advanced cancers, including colorectal, breast, bladder, sarcoma, and others. This trial established 14 mg/m2 as the MTD dose, as no significant cardiac or skin toxicity was observed with this dose and schedule.Citation23 The DLTs continued to be grade 3 thrombocytopenia and grade 3 fatigue, as seen in the earlier study. A romidepsin dose and schedule of 14 mg/m2 given on days 1, 8, and 15 of a 21-day cycle were established as the Phase II dose for further investigation.
Pharmacokinetic analysis was performed on 98 patients enrolled in the NCI-sponsored Phase II multicenter clinical trial of romidepsin in patients with CTCL in whom the drug was administered at 14 mg/m2 or 18 mg/m2 on day 1 during their first treatment cycle.Citation24 Using a noncompartmental analysis for the first dose, the mean half-life (2.95 hours) and observed clearance (9.6 h/m2) were comparable with previously reported parameters in the Phase I studies. After a 4-hour infusion, romidepsin was rapidly eliminated from the circulation with a short half-life of ∼3.5 hours, as observed in other studies. Owing to this rapid distribution and clearance, resulting in plasma concentrations below the limit of quantitation, terminal elimination rates (kel) could not be calculated. Romidepsin is extensively metabolized in vivo, primarily by the cytochromes P450 (CYP) 3A4, and to a lesser extent by the CYP3A5, and eliminated through bile and excreted in feces.Citation25,Citation26 Drug–drug interactions to be considered when administering romidepsin include any strong CYP3A4 or CYP3A5 inhibitors, such as ketoconazole, which has been shown to significantly lower romidepsin metabolism in vitro. Two-compartment population modeling was carried out to explore the effects of polymorphic variations in CYP3A4, CYP3A5, SLCO1B3, and ABCB1, all of which encode genes thought to be involved in the metabolism of romidepsin. Despite some minor interindividual variability, no statistical differences in romidepsin pharmacokinetic parameters were found between patients with wild-type genes and those with polymorphisms.
Pharmacodynamic studies have revealed the ability of serum from patients treated with romidepsin to induce cell cycle arrest and confirmed the histone acetylation increase in circulating peripheral blood mononuclear cells (PBMCs).Citation27 The maximal accumulation of acetyl H3 histones in PBMCs occurred at 4 hours after the end of the romidepsin infusion. Increased histone acetylation has also been reported in post-treatment biopsies. Other studies have also reported that in patients with acute myelogenous leukemia (AML) and CLL treated with 13 mg/m2 of romidepsin intravenously on days 1, 8, and 15, HDAC inhibition and histone acetylation of at least 100% were observed, and the levels of H3 and H4 histone deacetylation were higher at the 4-hour time point than 24 hours in the CLL and AML cell lysates. Additionally, an increase in p21 expression occurred concurrently with H4 histone acetylation of the p21 promoter.Citation28 In an attempt to find meaningful biomarkers of response to romidepsin, the molecular endpoints of romidepsin infusion were evaluated in 61 patients enrolled in the Phase II trial of romidepsin.Citation29 They were identified as laboratory parameters modified by romidepsin and included histone H3 acetylation and ABCB1 gene expression in PMBCs, ABCB1 gene expression in tumor biopsy samples, and blood fetal hemoglobin (HbF) levels, all of which were increased following romidepsin treatment. A somewhat lower proportion of patients demonstrated increased ABCB1 expression in PBMCs. When each surrogate was evaluated for association with either PK parameters or disease response, it was noted that the increase in histone acetylation measured in PBMCs at 24 hours correlated with Cmax and AUC and inversely correlated with clearance. In addition, histone acetylation in PBMCs at the 24-hour time point appeared to correlate with response, despite a lack of a statistically significant correlation between PK parameters and response. There was no correlation between the levels of ABCB1 induction and pharmacokinetic parameters, nor was there an association between induction of ABCB1 in biopsy specimens and disease response. In total, 60% of patients also had a greater than four-fold increase in circulating HbF detected in blood. Although this was correlated with response, it was also associated with increased time on study. These data suggest that for romidepsin, drug exposure dependent on drug clearance and drug activation are important determinants of response, consistent with data from other HDACis.
Clinical activity of romidepsin in T-cell lymphomas
The first Phase I clinical trial of romidepsin was conducted in 2001 at the NCI by Piekarz et al and included four patients with refractory T-cell lymphomas (two patients with SS, one patient with tumor-stage CTCL, and one patient with unspecified peripheral T-cell lymphoma).Citation30 The dosing schedule consisted of romidepsin administered at a dose of 12.7 mg/m2 or 17.8 mg/m2 as a 4-hour intravenous infusion on days 1 and 5 of a 21-day cycle, as described previously. To confirm biological activity, histone acetylation assays were performed on patients’ PBMCs. Responses were scored according to the World Health Organization criteria. Both patients with SS who were refractory to conventional chemotherapy agents had a rapid decrease in the percentage of circulating Sézary cells along with improvement in the skin erythema and edema following treatment with romidepsin. The first SS patient was removed from the study due to persistent methicillin-resistant Staphylococcus aureus infection, and the second SS patient was removed after five cycles because of erythema and pruritus. There was rapid clearing of all tumors after six cycles of treatment with romidepsin in the third patient, whereas the fourth patient with peripheral T-cell lymphoma was declared to be in complete remission after the eighth cycle of romidepsin, with complete disappearance of his skin lesions and diffuse adenopathy. At the end of this trial, it was concluded that romidepsin demonstrated significant clinical activity in the treatment of T-cell lymphomas and needed further investigation in a Phase II trial.
Based on the favorable responses observed in patients with CTCL during Phase I testing, a Phase II trial to evaluate the efficacy of romidepsin in these patients was initiated at the NCI and extended to other centers after initial encouraging observations.Citation31 A total of 71 patients with relapsed, refractory, or advanced CTCL with either MF or SS who had no more than two prior cytotoxic chemotherapy regimens were enrolled, although there was no restriction on the number of prior systemic biologic therapies such as steroids, retinoids, monoclonal antibodies, or topical therapies. A majority of the 71 patients had advanced disease (stage IIB to IVB) as follows: stage IVA (n = 28), stage IVB (n = 13), stage IIB (n = 15), stage IB (n = 6), stage IIIA and IIIB disease (n = 3), stage IIA (n = 2), and one patient alone had stage IA disease. The median number of prior therapies was four (0–14), including psoralen plus ultraviolet therapy, retinoids, radiation, total skin electron beam, photopheresis, interferon, and single or combination chemotherapy agents. Romidepsin was administered as a 4-hour intravenous infusion at the dose 14 mg/m2 on days 1, 8, and 15 of a 28-day schedule. The average number of cycles received by the patients was four (1–72), and the median number of doses was 12 (2–141). The response criterion used in this study was a composite score comprising the disease compartment that was involved, ie, skin, lymph nodes, or marrow. Disease in skin or viscera was assessed by Response Evaluation Criteria in Solid Tumors (RECIST) criteria,Citation32 lymph node involvement was assessed using International Working Group Guidelines (IWGG),Citation33 and bone marrow involvement was recorded as present or absent. Generalized erythroderma was scored as present or absent. According to the RECIST criteria, size of skin lesions was documented by color photography including a ruler. The overall response rate for the group of 71 patients was 34% (23%–46%) with four complete responses (7%) and 20 (28%) partial responses observed. Stable disease was noted in 26 patients (38%). The median duration of response was notable at 13.7 months (range 1–66+ months). Among the patients with a major response (complete or partial), the median time to response was 2 months (range 1–6 months). The median time to progression was 15.1 months for patients with a major response and 5.9 months for patients with stable disease. One of the striking features of romidepsin was the long duration of response noted that extended beyond 3 years, which was observed in some patients even after discontinuation of the drug. The complete responders included one patient with MF and three patients with SS. Among the 62 patients with stage IV disease, 18 patients (29%) achieved a complete or partial response. Overall, the drug was well tolerated, with toxicities similar to those observed in Phase I trials, such as fatigue (41%), nausea (52%), vomiting (20%), anorexia (21%), leukopenia (31%), granulocytopenia (37%), lymphopenia (21%), thrombocytopenia (39%), and anemia (37%). Hematologic abnormalities returned to baseline by the next cycle. Asymptomatic T-wave flattening (71%) or ST segment depression (9%) was observed, with no evidence of cardiac damage. Of note, a median increase of 14 ms in the QT interval was seen with 0.2% of >2000 electrocardiograms, demonstrating a QTcB (Bazett’s correction) interval of more than 500 ms.
One of the major drawbacks of this study was the tools used to assess response. The RECIST criteria for solid tumors and the IWGG for lymphomas used in this trial give inaccurate estimations of the disease burden in skin or blood. The Severity-Weighted Assessment Tool (SWAT) score developed by Stevens et alCitation34 uses the principle of grid-point counting and gives a more comprehensive, dynamic, and accurate measurement of the skin disease burden in MF.Citation35 This score represents the product of the percentage total body surface area (%TBSA) involvement of each lesion type (patch, plaque, and tumor or ulceration), multiplied by a weighting factor: SWAT = (patch %TBSA × 1) + (plaque %TBSA × 2) + (tumor or ulcer %TBSA × 3) within 12 body regions. The three weighted subtotals are added in order to determine the total score. It captures overall physical impressions of disease status on a continuous dimensionless numerical scale, thereby providing a defined, objective, and sensitive measure and hence is regarded as the best scoring system for skin disease in CTCL.
While the NCI study was being undertaken, Gloucester Pharmaceuticals started a parallel single-arm, open-label, prospective, Phase II trial of romidepsin in patients with pre-treated CTCL. It was a multicenter trial involving 33 centers in eight countries.Citation36 Ninety-six patients (59 with MF and 37 with SS) were enrolled in the study and were administered romidepsin at the dose of 14 mg/m2 as a 4-hour infusion on days 1, 8, and 15 of each 28-day cycle for up to six cycles with the option of extension of treatment in those with stable disease or response. Enrolled patients had the following characteristics: stage IVA (n = 24), stage III (n = 23), stage IIB (n = 21), stage IIA (n = 13), and stage IB (n = 15). The median number of prior therapies was four (1–11), which included skin-directed and systemic agents. This study employed a rigorous composite endpoint, which included the SWAT and erythroderma scores along with the RECIST criteria for the assessment of nodal disease and flow cytometry for changes in peripheral blood tumor burden to assess responses to romidepsin. Pruritus was reported monthly by the patients using a 100 mm visual analog scale (VAS).Citation37,Citation38 These various scores and scales were felt to be a better marker of overall meaningful tumor response. The primary endpoint of overall response rate was 34%, including six complete responses and 20 partial responses, with responses seen in all stages and all compartments of the disease (skin, blood, and lymph nodes). Of the 37 SS patients, 12 patients (32%) had a response to romidepsin, including two complete responses. A rapid, dramatic reduction in number of circulating Sézary cells was observed. There was a ≥50% reduction in SWAT and/or erythroderma scores in 38 patients (40%). The median duration of response was 15 months (range 0–19.8+ months) with a time to response of 2 months (range 0.9–4.8 months) and median time to complete response of 4 months (range 0.9–6.9 months). Ninety-two percent of the patients showed improvement in their pruritus score. Of note, clinically meaningful improvement in pruritus (assessed by VAS decrease of ≥30 mm or a score of 0 for 2 consecutive days) was observed in 28 (43%) patients. No serious adverse events were noted, with gastrointestinal intolerance and asthenia being the most common toxicities, and most of those were mild (grade 1 or 2). A clinically insignificant change in QTc interval was seen, which was attributed to antiemetics rather than to the drug per se.
These noteworthy results from two parallel, prospective, single-arm, multicenter trials were presented to the FDA and led to the approval of this agent in patients with advanced, refractory CTCL. The outcomes of major clinical trials of romidepsin as monotherapy have been summarized in .
Table 3 Summary of major clinical trials performed to date with romidepsin as a single agent
Toxicities of romidepsin that deserve attention
Cardiac toxicity associated with HDACi therapy has been the focus of attention in all clinical trials involving these agents and appears to be a class effect. When romidepsin was administered at the dose of 1 mg/kg or 2 mg/kg as a 4-hour intravenous infusion on days 1, 5, and 9 or 2 mg/kg as a 24-hour infusion on day 1 in Beagle dogs at the NCI, mortality occurred in all four dogs that received the 24-hour infusion and in one of the dogs that received the dose of 2 mg/kg over 4 hours. Lymphoid atrophy was noted in all three treatment groups.Citation20 This suggested efficacy of the 4-hour infusion and demonstrated toxicity of the 24-hour infusion. At very high doses, hemorrhages were detected in multiple organs, including the heart. Neither electrocardiogram abnormalities nor cardiac hemorrhages were observed when depsipeptide was infused over 4 hours. No cardiac lesions were detected in rats treated with very high doses.Citation21 Unpublished observations of QT interval prolongation and ST segment abnormalities were observed in the Beagle dogs treated with rapid infusions of romidepsin. This and similar asymptomatic electrocardiographic abnormalities noted in Phase I clinical trials involving a 4-hour infusion led to the incorporation of intensive cardiac monitoring in the Phase II trial for CTCL.Citation39 Cardiac studies such as serial electrocardiograms, cardiac enzymes, multiple gated acquisition scans, echocardiograms, baseline 24-hour Holter analysis, and telemetry monitoring were performed during the first dose of the first cycle. Cardiac studies from 282 cycles and 736 doses of romidepsin involving 2051 electrocardiograms and 161 left ventricular ejection fraction evaluations were reviewed. Although T-wave flattening (grade 1) and ST segment depression (grade 2) were observed in more than half of the electrocardiograms post-treatment, no impact on myocardial function was noted. Analysis of the QTc interval before and after treatment demonstrated a median QTc prolongation of 14 ms following treatment with romidepsin. Some potential explanations that have been proposed regarding the asymptomatic electrocardiogram abnormalities noted with romidepsin are pretreatment with serotonin inhibitors or concurrent use of medications known to cause QTc prolongation, electrolyte abnormalities of potassium and magnesium known to exist in patients with CTCL, and coexistence of comorbidities or pre-existing cardiac arrhythmias in the treatment population. Sudden death occurred in six patients enrolled in the Phase II trials of romidepsin. However, on retrospective analysis, all patients had cardiac comorbidities that predisposed them to risk of sudden death. With regard to cardiac dysfunction, clinical trials have excluded patients with pre-existing cardiac disease, long QT syndrome, or other risk factors for sudden death. Unless this class effect of HDACis is further investigated and more experience is gained with their use, it would be reasonable to adopt a safe strategy and avoid them in the patient population mentioned.
Other unexpected toxicities of romidepsin, including three cases of reactivation of latent DNA viruses, have been reported in the patients enrolled in the Phase II multi-institutional study of romidepsin in patients with T-cell lymphoma.Citation40 Two cases of Epstein–Barr virus (EBV) associated illness and one case of hepatitis B virus (HBV) reactivation were observed. The first patient with EBV reactivation died of multiorgan failure from disseminated EBV infection, and the second patient responded well to treatment with single-agent rituximab. The patient with HBV reactivation was started on treatment with single-agent entecavir with a favorable response. It was hypothesized that these complications were likely related to HDACi therapy through transcriptional activation of silenced promoters and induction of immunosuppression via depletion of circulating lymphocytes.
Nausea (all grades 52%–56%), asthenia (all grades 44%), anorexia (all grades 20%), vomiting (all grades 19%–26%), and aguesia (all grades 13%) were commonly seen in patients receiving romidepsin.Citation31,Citation36 Most of these adverse events were mild (grade 1 or 2). Only about 2%–7% of the patients experienced grade 3 or 4 toxicity, likely secondary to administration of serotonin inhibitors prior to and after romidepsin infusion.
Constitutional symptoms of fatigue (41%) and fever (25%) have been observed in patients on romidepsin treatment and felt to be cytokine mediated.Citation31 However, once again, 24% of the patients had grade 1 or 2 fatigue, and grade 3 or 4 was rare.
Leukopenia (all grades 30%), granulocytopenia (all grades 36%), thrombocytopenia (all grades 40%), and anemia (all grades 37%) were transient and rapidly reversible.Citation31 Furthermore, romidepsin does not affect the colony-forming units from bone marrow cells, and bone marrow biopsies performed in patients with romidepsin do not demonstrate significant myelosuppression.
Unanswered questions about the treatment of CTCL with romidepsin
The mechanism behind the efficacy of romidepsin and other HDACis as a class in T-cell lymphomas remains unclear. It is tempting to speculate that the responsive subset of T-cell lymphomas has its origin in an as yet unknown chromosomal arrangement that recruits the class I HDACis to the promoter of a gene that normally limits cell proliferation. However, such chromosomal aberrations have not been described in patients with T-cell lymphomas. Hence, it is possible that HDACis through their pleiotropic effects are merely regulating the balance between cell survival and apoptosis in patients with T-cell lymphomas similar to other disease entities rather than exerting efficacy via tumor-specific mechanisms. Better understanding of tumor biology in CTCL and cellular pathways affected by HDACis will be required to better define the role of these agents in CTCL, particularly in combination therapies. Why responses seen are far more common in hematologic malignancies than in solid tumors is also not understood.
Differences in the pharmacology, potency, and efficacy across HDACis are incompletely understood. Advanced pharmacodynamic studies with more clinical experience and incorporation of correlative studies into the trials across different tumors may answer some of these questions and expand the spectrum of activity of romidepsin.
Another pertinent question is the role of synergy between romidepsin and other chemotherapeutic agents in diseases such as CTCL. HDACis in combination with hypomethalating agents have shown clinical activity in early-phase clinical trials in myeloid malignancies.Citation41–Citation43 Romidepsin has also been investigated in combination with the proteasome inhibitor bortezomib in patients with relapsed and refractory multiple myeloma with encouraging results in a small Phase II trial of 22 patients.Citation44 The study showed good tolerability in the patients, with four complete responses, two very good partial responses, six partial responses, five minimal responses, and one stable disease. However, the role of this combination in CTCL remains to be explored. Not only will the choice of chemotherapeutic agent be an important area of research in such combination strategies but the sequence of administration will pose a greater challenge given the lack of clear understanding of the molecular effects of HDACis.
Given the plethora of HDACis now available for clinical use, molecular analysis of tumors for signatures that can predict sensitivity response to individual agents would be ideal. A retrospective analysis of pretreatment CTCL skin biopsies found that high nuclear STAT1 and phopho-STAT3 staining in lymphoma cells correlated with a lack of clinical response to vorinostat.Citation45 Although the status of STAT1 and phosphor-STAT3 as predictors of romidepsin response remains unknown, identification of such biomarkers that could predict the outcome of romidepsin in patients with CTCL should be a focus of future clinical trials.
Finally, given the promising activity of all HDACis in CTCL, it is important to compare them with one another either as single agents or in combination with other therapies in Phase III trials to see whether one drug emerges to have superior efficacy or use molecular analysis to tailor therapies for individual patients. Comparative efficacy, toxicity, and cost effectiveness between the two FDA-approved and indicated HDACi therapies romidepsin and vorinostat for CTCL have not been studied.
Identification of ways to exploit the use of romidepsin in solid tumors and combination with radiation will be needed to select cell types that are sensitive to the poorly understood epigenetic modifications induced by romidepsin.
Conclusion
Despite multiple unresolved questions with respect to the mechanism of action of romidepsin in CTCL and the fact that it is not a targeted therapy, romidepsin has demonstrated significant clinical activity in patients with refractory neoplasms such as MF and SS with good durability and tolerable side effects. We anticipate future trials comparing its role either as a single agent or in combination with other biologic agents in both frontline and relapsed settings. Designing and implementing randomized multicenter clinical trials with laboratory correlates to investigate the efficacy of drugs for rare diseases like CTCL will always be a challenge. Hence, it is imperative that insight into the biology of CTCL guides the development of novel agents.
Disclosure
The authors report no conflicts of interest in this work.
References
- CriscioneVDWeinstockMAIncidence of cutaneous T-cell lymphoma in the United States, 1973–2002Arch Dermatol200714385485917638728
- BunnPALambergSIReport of the Committee on Staging and Classification of Cutaneous T-cell LymphomasCancer Treat Rep197963725728445521
- VonderheidECBernengoMGThe Sezary Syndrome: hematologic criteriaHematol Oncol Clin North Am2003171367138914710890
- ZackheimHSAminSKashani-SabetMPrognosis in cutaneous T-cell lymphoma by skin stage: long term survival in 489 patientsJ Am Acad Dermatol19994041842510071312
- UedaHNakajimaHHoriYFR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968, I: Taxonomy, fermentation, isolation, physico-chemical and biological properties, and antitumor activityJ Antibiot (Tokyo)1994473013107513682
- UedaHMandaTMatsumotoSFR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968, III: Antitumor activities on experimental tumors in miceJ Antibiot (Tokyo)1994473153238175484
- NakajimaHKimYBTeranoHFR901228, a potent antitumor antibiotic, is a novel histone deacetylase inhibitorExp Cell Res19982411261339633520
- FurumaiRAkihisaMNobuyukiKFK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylasesCancer Res200262491612208741
- BhallaKNEpigenetic and chromatin modifiers as targeted therapy of hematologic malignanciesJ Clin Oncol2005233971399315897549
- PiekarzRBatesSEpigenetic modifiers: basic understanding and clinical developmentClin Cancer Res200915123918392619509169
- LeighEYanPGordonKSHistone deacetylase inhibitor panobinostat induced clinical responses with associated alterations in gene expression profiles in cutaneous T-cell lymphomaClin Cancer2008144500
- ByrdJCShinnCRaviRDepsipeptide (FR901228): a novel therapeutic agent with selective, in vitro activity against human B-cell chronic lymphocytic leukemia cellsBlood1999941401140810438728
- PiekarzRLRobeyRWZhanZT-cell lymphoma as a model for the use of histone deacetylase inhibitors in cancer therapy: impact of depsipeptide on molecular markers, therapeutic targets, and mechanisms of resistanceBlood20041034636464314996704
- KhanSBMaududiTBartonKAnalysis of histone deacetylase inhibitor, depsipeptide (FR901228), effect on multiple myelomaBr J Haematol200412515616115059137
- XiaoJAmyFPeterSEfflux of depsipeptide FK 228 (FR 901228, NSC-630176) is mediated by P-glycoprotein and multi drug resistantce-associated protein-1J Pharmacol Exp Ther20053326827615634944
- RobeyRWZhanZPiekarzRLIncreased MDRI expression in normal and malignant peripheral blood mononuclear cells obtained from patients receiving depsipeptide (FR901228, FK228, NSC630176)Clin Cancer Res2006121547155516533780
- BergSLStoneJXiaoJJPlasma and cerebrospinal fluid pharmacokinetics of depsipeptide (FR901228) in nonhuman primatesCancer Chemother Pharmacol200454858815042312
- PeartMJTaintonKMRuefliAANovel mechanisms of apoptosis induced by histone deacetylase inhibitorsClin Cancer Res2003634460
- NewboldALindermannRKCluseLACharacterization of the novel apoptotic and therapeutic activities of the histone deacetylase inhibitor romidepsinMol Cancer Ther200871066107918483296
- PageJGRodmanLEHeathJEEffect of infusion rate on toxicity of depsipeptide (NSC-630176)Proc Amer Assoc Cancer Res199536368
- LiZChanKKA subnanogram API LC/MS/MS quantitation method for depsipeptide FR901228 and its preclinical pharmacokineticsJ Pharm Biomed Anal200022334410727121
- SandorVBakkeSRobeyRPhase I clinical trial of histone deacetylase inhibitor, depsipeptide (FR 901228, NSC 630176) in patients with refractory neoplasmsClin Cancer Res2002871811895901
- MarshallJRizviNKauhJA phase I trial of depsipeptide (FR901228) in patients with advanced cancerJ Exp Ther Oncol2002232533212440223
- SukyungWErinRGXiaohongCPopulation pharmacokinetics of romidepsin in patients with cutaneous T-cell lymphoma and relapsed peripheral T-cell lymphomaClin Cancer Res2009151496150319228751
- ShiragaTTozukaZIshimuraRIdentification of cytochrome P450 enzymes involved in the metabolism of FK228, a potent histone deacetylase inhibitor, in human liver microsomesBiol Pharm Bull20052812412915635176
- FehrenbachTCuiYFaulstichHCharacterization of the transport of the bicyclic peptide phalloidin by human hepatic transport proteinsNauryn Schmiedebergs Archives Pharmacol2003368415420
- FouladiMFurmanWChinTPhase I study of depsipeptide in pediatric patients with refractory solid tumors: a Children’s Oncology Group reportJ Clin Oncol2006243678368516877737
- ByrdJCMarcuciGParthunMRA phase I and pharmacodynamic study of depsipeptide (FK228) in chronic lymphocytic leukemia and acute myeloid leukemiaBlood200510595996715466934
- SusanEBZhirongZKennethSLaboratory correlates for a phase II trial of romidepsin in cutaneous and peripheral T-cell lymphomaBr J Haematol200914825626719874311
- PiekarzRLRobeyRSandorVInhibitor of histone deacetylation, depsipeptide (FR901228), in the treatment of peripheral and cutaneous T-cell lymphoma: a case reportBlood2001982865286811675364
- PiekarzRFryeRTurnerMPhase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphomaJ Clin Oncol2009275410541719826128
- TherassePArbuckSGEisenhuerEANew guidelines to evaluate the response to treatment in solid tumors: European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of CanadaJ Natl Cancer Inst20009220521610655437
- ChesonBDHorningSJCoiffierBReport of an international work shop to standardize response criteria for non-Hodgkin’s lymphomas: NCI Sponsored International Working GroupJ Clin Oncol199917124410561185
- StevensSRKeMSParryEJQuantifying skin disease burden in mycosis fungoides-type cutaneous T-cell lymphomas: the Severity-Weighted Assessment Tool (SWAT)Arch Dermatol2002138424811790166
- HealdPClinical trials and efficacy assessment in the therapy of cutaneous T-cell lymphomaAnn N Y Acad Sci200194115516511594569
- WhittakerSDemierreMFKimEJFinal results from a multi-center, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphomaJ Clin Oncol201028294485449120697094
- OlsenEDuvicMFankelAPivotal phase III trial of two dose levels of denileukin diftitox for the treatment of cutaneous T-cell lymphomaJ Clin Oncol20011937638811208829
- EdelsonRBergerCGasparroFTreatment of cutaneous T-cell lymphoma by extracorporeal photochemotherapy: preliminary resultsN Engl J Med19873162973033543674
- PiekarzRFryeRWrightJCardiac studies in patients with depsipeptide, FK28, in a phase II trial for T-cell lymphomaClin Cancer Res2006123762377316778104
- RitchieDPiekarzRBlomberyPReactivation of DNA viruses in association with histone deacetylase inhibitor therapy: a case series reportHaematologica2009941618162219608677
- MaslakPChanelSCamachoLHPilot study of combination transcriptional modulation therapy with sodium phenylbutyrate and 5-azacytidine in patients with acute myeloid leukemia or myelodysplastic syndromeLeukemia20062021221716357841
- BlumWKlisovicRBHackansonBPhase I study of decitabine alone or in combination with valproic acid in acute myeloid leukemiaJ Clin Oncol2007253884389117679729
- SorianoAOYanhHFaderlSSafety and clinical activity of the combination of 5-azacytidine, valproic acid, and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndromeBlood20071102302230817596541
- BerensonJRYellinOMapesPA phase II study of a 1-hour infusion of romidepsin combined with bortezomib for multiple myeloma (MM) patients with relapsed or refractory diseaseJ Clin Oncol200927e19508
- JFantinVRLobodaAPaweletzCPConstitutive activation of signal transducers and activators of transcription predicts vorinostat resistance in cutaneous T-cell lymphomaCancer Res2008683785379418483262
- OlsenEAKimYHKuzelTMPhase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphomaJ Clin Oncol2007253109311517577020
- DuvicMBeckerJCDalleSPhase II trial of oral panobinostat (LBH589) in patients with refractory cutaneous T-cell lymphoma (CTCL)Blood20081121005101118477770
- FossFAdvaniRHymesKActivity of belinostat in patients with recurrent or refractory peripheral or cutaneous T-cell lymphomaHaematol Meet Rep2009313940
- ParkerCMolifeRKaravasilisVRomidepsin (FK228), a histone deacetylase inhibitor: Final results of a phase II study in metastatic hormone refractory prostate cancer (HRPC)J Clin Oncol, ASCO Annual Meeting Proceedings2007518S15507
- StadlerWMMargolinKFerberSA phase II study of depsipeptide in refractory metastatic renal cell cancerClin Genitourin Cancer200651576016859580
- WhiteheadRPRankinCHoffPMPhase II trial of romidepsin (NSC-630176) in previously treated colorectal cancer patients with advanced disease: a Southwest Oncology Group study (S0336)Invest New Drugs200927546947518941712
- NiesvizkyRElySMarkTPhase 2 trial of the histone deacetylase inhibitor romidepsin for the treatment of refractory multiple myelomaCancer2011117233634220862746