Abstract
Background
Several micro-environmental and cell-intrinsic stimuli cause tumor cells to undergo endoplasmic reticulum (ER) stress in vivo. The occurrence of an ER stress response has been associated with tumor progression and angiogenesis. Recently, we found that pharmacological induction of ER stress in B lymphoma cells upregulates the transcription of several pro-inflammatory cytokines.
Results
Here, we show that transgenic adenocarcinoma of the mouse prostate (TRAMP) C1 murine prostate cancer cells induced to undergo ER stress in vitro activate the transcription of interleukin 6 (IL-6), interleukin 23p19 (IL-23p19), and tumor necrosis factor α (TNF-α). Furthermore we show that TRAMP C1 tumors growing in vivo spontaneously experience ER stress and that transcription of IL-6, IL-23p19, and TNF-α correlates with the in vivo ER stress response.
Conclusions
These results suggest that an ER stress response in prostate cancer cells activates a program of pro-inflammatory cytokine transcription. A possible implication of this finding is that cancer cells may use the ER stress response to modify their microenvironment.
Background
The endoplasmic reticulum (ER) is the initial checkpoint for the biosynthesis, folding, assembly, and modification of membrane-bound and secreted proteins in all eukaryotic cells. Stimuli that cause the accumulation of un/misfolded proteins in the ER lumen result in a condition known as ER stress.Citation1 Eukaryotic cells have evolved a set of intracellular signaling pathways known collectively as the unfolded protein response (UPR) that facilitates cellular adaptation to ER stress. In mammalian cells, the UPR is initiated by three ER membrane-bound sensors (inositol-requiring protein 1α (Ire1α), Activating transcription factor 6 (Atf6), protein kinase-like endoplasmic reticulum kinase (PERK)), which in unstressed cells, are maintained in an inactive state through association with the ER chaperone molecule Grp78.Citation2 When a cell experiences ER stress, Grp78 disassociates from each of the three sensor molecules to preferentially bind un/misfolded proteins, causing each sensor to activate downstream signaling cascades, which ameliorate ER stress via several mechanisms, including selective translation inhibition and upregulation of genes that encode enzymes that aid in protein folding, maturation, and degradation.Citation3 Involved in this homeostatic/regulatory cascade are two target genes Myd116 (growth arrest and DNA damage-inducible protein (Gadd34)) and C/EBP homologous protein (CHOP) that are associated with translational recovery and apoptosis, respectively.Citation1
Tumor cells in vivo are continuously exposed to ER stress in their microenvironment through hypoxia, low nutrient supply, and low pH. Tumor-intrinsic causes of ER stress include oxidative stress, defective glycosylation, and defects in calcium homeostasis.Citation4 Evidence suggests that the ability to mount the UPR confers upon tumors a growth advantage. Primary human tumor cells of many different origins, including breast,Citation5 lung,Citation6 liver,Citation7 colon,Citation8 and prostate,Citation9 have been shown to upregulate various elements of the ER stress response, including GRP78. In primary human melanoma specimens, the level of GRP78 positively correlates with tumor progression.Citation10 Conversely, Grp78 hemizygous mice crossed with MMTVPyVT heterozygous transgenic mice display significantly decreased tumor proliferation, survival, and angiogenesis compared to Grp78+/+, PyT mice.Citation11 Additionally, the inactivation of ER stress signaling by mutations of PERK, or by the introduction of a dominant-negative PERK, in human colon cancer cells, results in tumors that are smaller, grow less rapidly, and display abnormal angiogenic ability, as compared to their normal counterparts when implanted into mice.Citation12,Citation13
Since Virchow’s original suggestion of a link between inflammation and tumorigenesis, the idea that inflammation in the tumor microenvironment serves as a potent driver of tumor progression has been validated by epidemiological and molecular evidence. For instance, gastrointestinal carcinogenesis is associated with Helicobacter pylori infection, and lung cancer correlates with exposure to smoking and asbestos.Citation14,Citation15 Tumor necrosis factor α (TNF-α) produced by stromal cells causes adjacent hepatocytes to undergo transformation into malignant cells via nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activationCitation16 and, conversely, deletion of NF-κB in hepatocytes reduces the incidence of liver tumors.Citation17 An additional source of inflammation in the tumor microenvironment is infiltrating leukocytes, most notably, tumor-associated macrophages.Citation18–Citation20
Recently, ER stress has been linked both to several inflammatory diseases and cancer.Citation3,Citation4 Support for the idea that ER stress signaling activates an inflammatory program comes from evidence demonstrating that signaling through each of the three ER stress sensors can activate NF-κB, a master regulator of inflammation.Citation21–Citation23 Previous work from this laboratory suggested a link between ER stress and the transcription of pro-inflammatory cytokines in vitro. Notably, genome-wide array of RNA of murine B lymphoma cells subjected to ER stress induced by thapsigargin, a canonical ER stressor, showed increased transcription of pro-inflammatory cytokines, including interleukin (IL)-6, IL-23p19, and TNF-α.Citation24 Here we present evidence that prostate cancer cells undergoing ER stress in vitro or in vivo also activate a program of proinflammatory cytokine transcription.
Results and discussion
We used quantitative PCR (qPCR) to analyze the effect of thapsigargin on murine transgenic adenocarcinoma of the mouse prostate (TRAMP) C1 prostate cancer cells in vitro. As expected, cells exposed to thapsigargin (0.3 μM) upregulated the ER stress-responsive genes Grp78, Gadd34, and CHOP relative to vehicle-treated cells (). IL-6 and IL-23p19 transcript levels also increased 26- and 53-fold, respectively, over vehicle-treated cells at the latest time point measured (). TNF-α transcript increased at 8 hours and declined thereafter. Increased transcription of TNF-α was observed but its kinetics in vitro seems to follow a pattern inverse to that of Grp78, Gadd34, and CHOP (, data not shown), suggesting that it may be regulated differently than IL-6 and IL-23p19 by UPR signaling.
Figure 1 TRAMP C1 cells activate pro-tumor inflammatory cytokines during ER stress in vitro. TRAMP C1 cells were treated with thapsigargin (Tg) or vehicle control for the indicated times and assayed for (a) ER stress marker expression, and (b) pro-inflammatory cytokine transcription, by qPCR. Data columns indicate the mean fold difference in transcript level between Tg- and vehicle-treated TRAMP C1 cells. Error bars represent standard error of the mean (sem.) of 2 independent experiments.
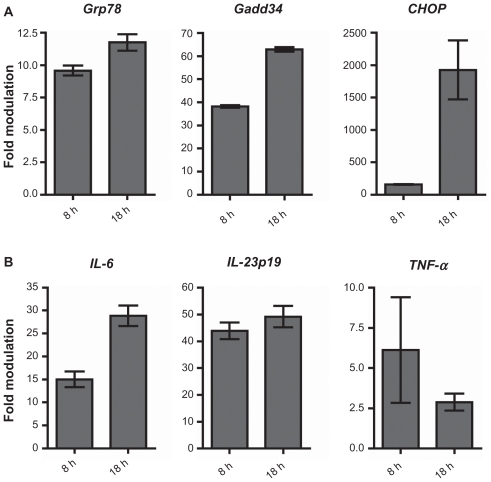
Corroborating these data, we observed an increase in intracellular IL-6 protein in TRAMP-C1 cells treated for 18 hours with thapsigargin (data not shown). Taken together, these data suggest that induction of ER stress in prostate cancer cells activates a program of pro-inflammatory cytokine transcription. It should be pointed out that we observed the same phenomenon in diverse tumor cell lines representative of melanoma, ovarian cancer, and T- and B-cell lymphoma (Gonzalo Almanza, unpublished data).
To corroborate this notion and assess whether a similar phenomenon would occur spontaneously in vivo, we utilized a heterotopic tumor transplant model. Briefly, 5 × 106 TRAMP C1 cells were injected subcutaneously into syngeneic C57BL/6 mice, and the tumors excised 7 days later to assess the activation of ER stress markers and cytokine transcription by qPCR. All but one tumor showed increased expression of the ER stress marker Grp78 (data not shown) and tumor IL-6, IL-23p19, and TNF-α expression highly correlated with Grp78 expression (). These data suggest that TRAMP C1 tumors undergo spontaneous ER stress in vivo and upregulate the transcription of pro-inflammatory cytokine genes.
Figure 2 TRAMP C1 cells forming tumors in vivo undergo ER stress and transcriptional activation of pro-inflammatory cytokine genes. TRAMP C1 cells (5 × 106) were injected subcutaneously into 12- to 14-week-old male C57BL/6 mice. Seven days after injection, tumors were surgically excised, mechanically disassociated, and assayed for Grp78 and pro-inflammatory cytokine transcription, by qPCR. Data points refer to individual tumors, and indicate the fold modulation in transcript level between tumor samples and spleen cells from tumor-bearing mice. Correlation was sought using a two-tailed Pearson correlation test. *p = 0.05, **p < 0.05.
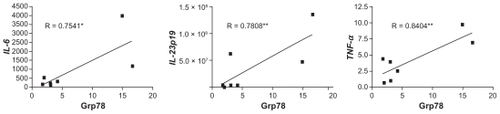
Admittedly, the exact source of these cytokines was not determined and is presently unknown. However, since in vitro cultured TRAMP C1 cells activate the transcription of IL-6, IL-23p19, and TNF-α under ER stress, we argue that ER-stressed tumor cells are a likely source of these cytokines in vivo. A contribution by tumor-associated myeloid cellsCitation25 to the observed transcriptional activation of IL-6, IL-23p19, and TNF-α cannot be formally excluded; however, since the accumulation of myeloid cells in tumors on day 7 post- implantation (as was the case in this study) is negligible, if any,Citation26 we maintain that the present results can be explained on the basis of tumor cells being the source of pro-inflammatory cytokine transcription. Future experiments based on cell sorting, coupled with phenotype characterization, will be needed to precisely assess the role of other cells in tumors in vivo.
The tumorigenic nature of IL-6, IL-23, and TNF-αCitation27–Citation29 as well as of other molecules released by tumor cells and tumor infiltrating immune cells under ER stress,Citation30 makes it plausible to suggest that one of the mechanisms by which ER stress aids tumor growth is by mediating the release of these molecules, which in turn shape the immune landscape of the microenvironment to favor tumor progression. It will be interesting to see whether tumor cells under ER stress can influence surrounding immune cells, such as macrophages. In light of the fact that IL-6 and IL-23 have been shown to be involved in the differentiation and maintenance of the Th17 lineage,Citation31 it is also possible that ER stress mediated IL-6 and IL-23 production by tumor cells may enable them to modulate CD4+ T-cells in their microenvironment.
Methods
Cells
TRAMP C1 cells were originally obtained through the courtesy of Dr Andrew Weinberg (Providence Portland Medical Center) and grown in complete RPMI-1640 medium supplemented with 10% fetal calf serum. Cells were treated with thapsigargin (Alexis Biochemicals) initially dissolved in 100% ethanol and diluted in culture medium to a final concentration of 0.3 μM, or an equal volume of vehicle only, for the indicated times.
Mice and in vivo experiments
C57BL/6 Mice were purchased from Charles River and housed at the Moores Cancer Center Animal Facility and handled in accordance with University of California, San Diego Animal Subjects Program Guidelines (San Diego, CA, USA). For tumor inoculation, 5 × 106 TRAMP C1 cells were injected subcutaneously into the flank of 12–14 week old, male, wild-type C57BL/6 mice. Mice were sacrificed 7 days after tumor injection when ~4 mm tumors were visible. Tumors were surgically excised and mechanically dispersed into cell suspensions. Spleen cells from tumor bearing mice C57BL/6 mice were similarly prepared and used as controls.
Quantitative RT-PCR
RNA was isolated from cell suspensions using the Nucleopsin RNA II Kit (Macherey-Nagel). Genomic DNA was digested by on-column treatment with DNase. Concentration and purity of RNA was determined by analysis on a NanoDrop spectrophotometer (Thermo Scientific). cDNA was obtained using the High Capacity cDNA Synthesis kit (Applied Bio-systems) and quantitative PCR was performed in triplicate on an ABI StepOne system using TaqMan reagents. Target gene expression was normalized to β-actin, and analyzed using the – ΔΔCt relative quantification method. Validated FAM-labeled mouse IL-23p19, IL-6, TNF-α, Ddit3 (CHOP), Myd116 (Gadd34), Hspa5 (Grp78), and VIC-labeled mouse β-actin TaqMan primer/probe sets (Applied Biosystems) were used.
Statistical analysis
Statistical analysis was performed using two-tailed Pearson correlation test with 95% confidence with the aid of Graph- Pad Prism software (GraphPad Software, Inc., California, USA).
Conclusion
This report shows that murine prostate cancer cells undergoing ER stress initiate a program of pro-inflammatory cytokine transcription. TRAMP C1 cells treated in vitro with a canonical inducer of ER stress upregulate transcription of IL-23p19 and IL-6 concomitantly with upregulation of ER stress markers. It also shows that TRAMP C1 tumors in vivo spontaneously upregulate markers of ER stress which is strongly correlated with the transcription of IL-6, IL-23p19, and TNF-α.
Acknowledgment
The authors are thankful to Dr Xochitl Cortez-Gonzalez for helpful discussion.
Disclosure
The authors report no conflicts of interest in this work.
References
- HardingHPCalfonMUranoFTranscriptional and translational control in the Mammalian unfolded protein responseAnnu Rev Cell Dev Biol20021857559912142265
- SchroderMKaufmanRJER stress and the unfolded protein responseMutat Res2005569296315603751
- ZhangKKaufmanRJFrom endoplasmic-reticulum stress to the inflammatory responseNature200845445546218650916
- MaYHendershotLMThe role of the unfolded protein response in tumour development: friend or foeNat Rev Cancer2004496697715573118
- FernandezPMTabbaraSOJacobsLKOverexpression of the glucose-regulated stress gene GRP78 in malignant but not benign human breast lesionsBreast Cancer Res Treat200059152610752676
- UramotoHSugioKOyamaTExpression of endoplasmic reticulum molecular chaperone Grp78 in human lung cancer and its clinical significanceLung Cancer200549556215949590
- ShudaMKondohNImazekiNActivation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesisJ Hepatol20033860561412713871
- XingXLaiMWangYOverexpression of glucose-regulated protein 78 in colon cancerClin Chim Acta200636430831516182273
- DaneshmandSQuekMLLinEGlucose-regulated protein GRP78 is up-regulated in prostate cancer and correlates with recurrence and survivalHum Pathol2007381547155217640713
- ZhuangLScolyerRALeeCSExpression of glucose-regulated stress protein GRP78 is related to progression of melanomaHistopathology20095446247019309398
- DongDNiMLiJCritical role of the stress chaperone GRP78/ BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor developmentCancer Res20086849850518199545
- AcharyaMRSparreboomAVenitzJFiggWDRational development of histone deacetylase inhibitors as anticancer agents: a reviewMol Pharmacol20056891793215955865
- BlaisJDAddisonCLEdgeRPerk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stressMol Cell Biol2006269517953217030613
- BalkwillFMantovaniAInflammation and cancer: back to VirchowLancet200135753954511229684
- SchwartsburdPMChronic inflammation as inductor of pro-cancer microenvironment: pathogenesis of dysregulated feedback controlCancer Metastasis Rev2003229510212716041
- PikarskyEPoratRMSteinINF-kappaB functions as a tumour promoter in inflammation-associated cancerNature200443146146615329734
- LuoJLMaedaSHsuLCInhibition of NF-kappaB in cancer cells converts inflammation- induced tumor growth mediated by TNFalpha to TRAIL-mediated tumor regressionCancer Cell2004629730515380520
- ErroiASironiMChiaffarinoFIL-1 and IL-6 release by tumor-associated macrophages from human ovarian carcinomaInt J Cancer1989447958012583859
- GretenFREckmannLGretenTFIKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancerCell200411828529615294155
- KortylewskiMXinHKujawskiMRegulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironmentCancer Cell20091511412319185846
- KanekoMNiinumaYNomuraYActivation signal of nuclear factor-kappa B in response to endoplasmic reticulum stress is transduced via IRE1 and tumor necrosis factor receptor-associated factor 2Biol Pharm Bull20032693193512843613
- DengJLuPDZhangYTranslational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2Mol Cell Biol200424101611016815542827
- KitamuraMBiphasic, Bidirectional Regulation of NF-kappaB by Endoplasmic Reticulum StressAntioxid Redox Signal2009112353236419187000
- WheelerMCRizziMSasikRKDEL-Retained Antigen in B Lymphocytes Induces a Proinflammatory Response: A Possible Role for Endoplasmic Reticulum Stress in Adaptive T Cell ImmunityJ Immunol200818125626418566391
- MantovaniASicaAAllavenaPTumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activationHum Immunol20097032533019236898
- YangLDeBuskLMFukudaKExpansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesisCancer Cell2004640942115488763
- NauglerWEKarinMThe wolf in sheep’s clothing: the role of interleukin-6 in immunity, inflammation and cancerTrends Mol Med20081410911918261959
- MummJBOftMCytokine-based transformation of immune surveillance into tumor-promoting inflammationOncogene2008275913591918836472
- LangowskiJLZhangXWuLIL-23 promotes tumour incidence and growthNature200644246146516688182
- FlowersMTKellerMPChoiYLiver gene expression analysis reveals endoplasmic reticulum stress and metabolic dysfunction in SCD1-deficient mice fed a very low-fat dietPhysiol Genomics20083336137218381840
- KornTOukkaMKuchrooVBettelliETh17 cells: effector T cells with inflammatory propertiesSemin Immunol20071936237118035554