
Abstract
Inflammation is triggered by a repertoire of receptors detecting infections and damages. Some of these receptors directly bind microbial ligands, while others recognize endogenous molecules exposed under stress conditions, including infections. Most of these receptors can be engaged by a relatively limited number of stimuli. Differently, NLRP3 acts as a broad sensor of cell homeostasis rupture and can be activated downstream of a plethora of stimuli. NLRP3 then assembles a multiprotein platform resulting in caspase-1 activation, which controls, by direct cleavage, the maturation of cytosolic pro-cytokines including pro-interleukin-1β. In addition, caspase-1 processes cytosolic gasdermin-D and unleashes its pore-forming N-terminal domain, leading to the release of mature cytosolic cytokines and alarmins, as well as pyroptotic cell lysis. Accumulating evidences of the aggravating role of NLRP3-mediated inflammation in various highly prevalent human conditions, including diabetes, neurodegenerative and cardiovascular diseases, raises a huge clinical interest. Nevertheless, the molecular mechanism governing NLRP3 activation remains insufficiently understood. In line with the detrimental consequences of NLRP3 activation illustrated by the aforementioned pathologies, this process is tightly regulated. In this review, we address the current understanding of the control of NLRP3 activity which can be divided into two coordinated processes referred to as priming and activation. In particular, we detail the emerging role of NLRP3 post-translational modifications critical in inflammasome assembly regulation.
Introduction
Inflammation is usually triggered in response to infections and/or tissue damages. It constitutes a protective response, critical for the recruitment of immune cells mediating pathogen clearance and tissue repair at the affected site.Citation1 Pathogens and damages are sensed by tissue resident cells, including innate immune cells, through a repertoire of germline-encoded dedicated receptors known as pattern recognition receptors (PRRs). These receptors are activated by pathogen-associated molecular patterns (PAMPs), typically shared throughout large kindred of microbes but absent from the host organism, and by host-derived cellular stress signals, markers of tissue and cell homeostasis disruption, known as damage-associated molecular patterns (DAMPs).Citation2,Citation3
Many inflammatory cytokines such as interleukin-6 (IL-6), tumor necrosis factor (TNF), and interferon-β (IFN-β) are regulated at the transcriptional level and secreted through the endoplasmic reticulum (ER)/Golgi pathway. Others, including IL-1β and IL-18, are produced as cytosolic precursors and their secretion is tightly controlled by inflammatory caspases, namely caspase-1 and -11 in mice, and caspase-1, -4, and -5 in humans. These caspases are activated within cytosolic multimolecular complexes named inflammasomes (). Canonical inflammasomes form activation platforms for caspase-1. Their assembly depends on some dedicated cytosolic PRRs from the nucleotide-binding domain leucin-rich repeat (NLR) family including NLR and pyrin domain containing receptor 1 (NLRP1), NLRP3, and NLR and caspase recruitment domain containing receptor 4 (NLRC4); the AIM2-like receptors (ALR) family including absent in melanoma 2 (AIM2); or the tripartite motif (TRIM) family including pyrin. Activation of these PRRs triggers either their oligomerization (as for NLRC4 or AIM2)Citation4,Citation5 or conformational changes of pre-formed oligomers (as for NLRP3).Citation6 The PRR oligomer then recruits the adaptor protein ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain [CARD]) through pyrin domain (PYD)–PYD interactions and constitutes a nucleation platform for filaments of ASC. The surface of the filament in turn enables the nucleation of filaments of the effector pro-caspase-1 through CARD–CARD interactions.Citation7 Inflammasomes form specks, also referred to as pyroptosomes, in the micron range in size and unique per cell, that can be observed by light microscopy.Citation8 Pyroptosome assembly constitutes an all-or-nothing response at the single cell level. Noteworthy, NLRC4 which possesses a CARD domain can either directly recruit pro-caspase-1 or trigger the formation of ASC filaments, serving as an amplification mechanism required for cytokine maturation but dispensable for pyroptosis.Citation9 The inflammasomes that control the activation of caspase-11 in mice and caspase-4 and -5 in human in response to cytosolic lipopolysaccharide (LPS) are referred to as non-canonical. Their composition remains poorly understood, and the lipid A moiety of LPS has been proposed to directly interact with the CARD of caspase-11, -4, and -5 which then undergo oligomerization, resulting in the activation of the caspases.Citation10 Therefore, non-canonical inflammasome assembly does not depend on dedicated PRRs, and caspase-11, -4, and -5 act as receptors themselves.
Figure 1 Canonical and non-canonical inflammasomes.
Notes: Canonical inflammasomes assemble downstream of cytosolic PRRs including NLRP1, NLRP3, NLRC4, AIM2, and pyrin and control the activation of caspase-1. The ASC adaptor forms multimeric filaments and amplifies the response. The non-canonical inflammasomes are activated by cytosolic LPS and result in caspase-4 and -5 (homologs of mouse caspase-11) activation. All these inflammatory caspases cleave GSDMD which then forms cytolytic pores leading to pyroptosis. In addition, caspase-1 controls the maturation of cytosolic pro-cytokines.
Abbreviations: PRRs, pattern recognition receptors; ASC, apoptosis-associated speck-like protein containing a CARD domain; LPS, lipopolysaccharide; GSDMD, gasdermin-D; h, human; m, mouse.
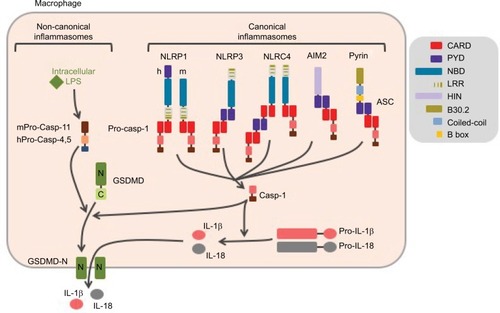
The caspase-1 protease controls maturation of pro-IL-1β and pro-IL-18, produced as pro-cytokines in the cytosol by direct cleavage of the pro-forms.Citation11 In addition, all inflammatory caspases trigger a pro-inflammatory form of cell death named pyroptosis (from Greek roots pyro, relating to fire or fever, and ptosis, relating to falling). This process is mediated by the cleavage of the cytoplasmic gasdermin-D (GSDMD) protein, which leads to the release of its N-terminal domain that oligomerizes as pores in the plasma membrane.Citation12–Citation14 GSDMD pores and cell lysis mediate the release of mature IL-1β and IL-18 together with IL-1a and many other alarm-ins, as well as the ASC speck. Extracellular ASC specks enter neighbor cells by phagocytosis and propagate the inflammatory signal.Citation15,Citation16 Noteworthy, caspase-11, -4, and -5 do not directly cleave pro-IL-1β and pro-IL-18. Non-canonical inflammasome controls cytokine maturation through sub-lytic plasma membrane permeabilization by GSDMD pores that activate the NLRP3 inflammasome.Citation12,Citation17
NLRP3, also known as cryopyrin, CIAS1, or NALP3, was initially described for its genetic association with forms of hereditary autoinflammation known as familial cold autoinflam-mation syndrome (FCAS), Muckle–Wells syndrome (MWS), and neonatal-onset multisystem inflammatory disease/chronic infantile neurologic cutaneous articular syndrome (NOMID/CINCA), in the order of increasing severity.Citation18,Citation19 These diseases were then collectively named cryopyrin-associated periodic syndromes (CAPS). Since then, NLRP3-mediated inflammation has been additionally recognized to fuel a number of multifactorial diseases including gouty arthritis, atherosclerosis, type 2 diabetes, and non-alcoholic steatohepatitis, as well as several neurodegenerative diseases, including amyotrophic lateral sclerosis, Alzheimer’s and Parkinson’s diseases, and other aging-related diseases.Citation20–Citation23 NLRP3 has also been involved in the response against diverse pathogens including ameba, protozoans including Plasmodium, fungi including Candida albicans and Saccharomyces cerevisiae, bacteria including Staphylococcus aureus and Listeria monocytogenes, and viruses including influenza, Sendai, and adenoviruses.Citation24 On contrary to the abovementioned conditions, NLRP3-dependent inflammation is mostly protective against infections, although the impact of Nlrp3 deficiency is usually limited, probably due to some redundancies among PRRs. NLRP3 is a 115 kDa cytosolic protein composed of three domains: a C-terminal leucin-rich repeat (LRR) with poorly characterized regulatory functions, a central oligomerization domain NACHT or nucleotide binding oligomerization domain (NOD) with an ATPase activity, and a N-terminal pyrin domain (PYD) that recruits ASC.Citation18 NLRP3 is activated by numerous and highly diverse PAMPs/DAMPs as, for example, hyperglycemia, ceramides, fatty acids, protein aggregates (β-amyloid, islet amyloid polypeptide), crystals (cholesterol, sodium urate, asbestos, silica, calcium pyrophosphate, alum), extracellular ATP, hyaluronic acid, pore-forming bacterial toxins including nigericin, bacterial and viral nucleic acids, and hemozoin.Citation25–Citation27 The great diversity in the structures of these activators excludes the fact that NLRP3 acts as their direct receptor. Instead, it is now well accepted that NLRP3 indirectly senses cytosolic stress. In this review, we describe the recent advances in our understanding of signaling pathways controlling NLRP3 activation, with a particular focus on the newly discovered key role of NLRP3 post-translational modifications in inflammasome assembly.
Signaling pathways controlling NLRP3 inflammasome assembly
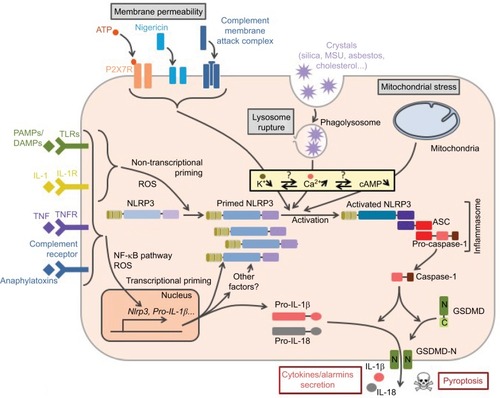
Regulation of the NLRP3 inflammasome has been most extensively investigated in macrophages. In these cells, NLRP3 inflammasome assembly results from a two-step process referred to as priming and activation (). The priming signal usually corresponds to cytokines or DAMPs/PAMPs recognized by receptors that transcriptionally control the expression of numerous genes. The activation signal can be provided by various cellular stresses and is transcription independent.
Figure 2 Control of NLRP3 inflammasome assembly.
Notes: Inflammasome formation requires coordinated NLRP3 priming and activation. PAMPs/DAMPs and cytokines prime NLRP3 through rapid post-translational modifications (non-transcriptional priming) and then NF-κB-dependent transcriptional upregulation of NLRP3 and putative other factors (transcriptional priming). Activation signals are provided by various stress signals including change in plasma membrane permeability, lysosome rupture, and metabolic stress. Downstream signaling pathways rely on cytosolic K+ decrease. Increase in cytosolic Ca2+ and decrease in cAMP are also often observed. NLRP3 activation results in inflammasome assembly and caspase-1 activation.
Abbreviations: PAMPs, pathogen-associated molecular patterns; DAMPs, damage-associated molecular patterns; TLR, Toll-like receptor; TNFR, tumor necrosis factor receptor; MSU, monosodium urate; ROS, reactive oxygen species; GSDMD, gasdermin-D; ASC, apoptosis-associated speck-like protein containing a CARD domain.
NLRP3 inflammasome priming
Consistent with the high diversity of the conditions leading to NLRP3 inflammasome assembly, a large number of receptors, including PRRs as well as cytokine and anaphylatoxin receptors, control NLRP3 priming. Among the PRRs, Toll-like receptors (TLRs), nucleotide-binding oligomerization domain 2 (NOD2), and receptor for advanced glycation endproducts (RAGE) can prime NLRP3 for further activation.Citation28–Citation32 The immune system itself can autonomously prime NLRP3 through the anaphylatoxin C3a and C5a receptors and cytokines IL-1 and TNF receptors.Citation33–Citation36 In addition, excessive reactive oxygen species (ROS) production also leads to NLRP3 priming.
Upon TLR binding by their agonists, several pathways resulting in NLRP3 priming are successively activated over time (). The first pathway that has been described relies on transcription activation downstream of these receptors and de novo protein synthesis.Citation35 This pathway is referred to as transcriptional priming or late priming, since it requires several hours (>3 h) before NLRP3 is licensed for further activation. Transcriptional priming depends on myeloid differentiation primary response 88 (MyD88), TIR domain-containing adapter-inducing interferon-β (TRIF), Fas-associated protein with death domain (FADD), and caspase-8, as well as ROS, which activate nuclear factor-κB (NF-κB).Citation37–Citation39 Indeed, while NLRP3 expression is low and the major caspase-1 substrate pro-IL-1β is not expressed in resting macrophages, both are highly transcriptionally upregulated downstream of TLR activation.Citation35 In addition, experimental overexpression of NLRP3 alleviates priming requirement for further NLRP3 activation, suggesting that priming mechanism may rely on this upregulation of NLRP3 expression level.Citation35,Citation39,Citation40 Expressions of ASC, pro-caspase-1, and other substrates, including IL-18, are constitutive and are not affected upon priming in cultured macrophages.
Figure 3 NLRP3 priming.
Notes: Several pathways successively control NLRP3 priming following TLR activation. The early and intermediate non-transcriptional pathways are independent of de novo protein synthesis and rely on post-translational modifications. They are, respectively, engaged by the MyD88 and the TRIF adaptors. Prolonged TLR or cytokine receptor activation prime NLRP3 through NF-κB-dependent transcriptional upregulation of NLRP3 and putative other factors. Both MyD88- and TRIF-dependent signaling drives this late transcriptional priming. All priming pathways depend on mitochondrial ROS.
Abbreviations: TLR, Toll-like receptor; ROS, reactive oxygen species; LPS, lipopolysaccharide.
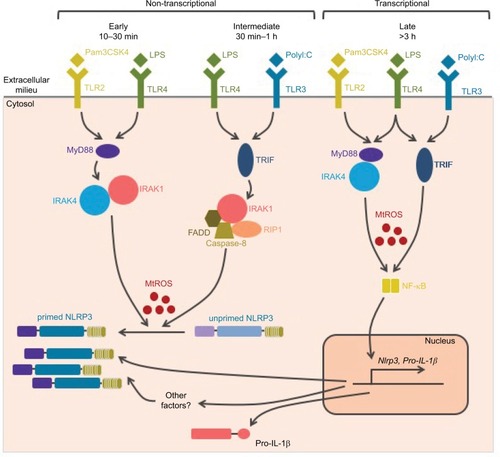
More recently, accumulative evidence highlighted a transcriptional-independent priming mechanism. Indeed, TLR agonists, but not IL-1β or TNF cytokines, license very quickly and transiently NLRP3 for further activation independently of protein synthesis.Citation36,Citation41–Citation44 These reports imply that NLRP3 basal expression level may be sufficient for the inflammasome assembly and that transcriptional priming may rely on the expression of other factors that remain to be identified. In addition, these observations suggest that several priming mechanisms may exist, and that early after the activation of TLRs, priming may rely solely on post-translational modifications. This non-transcriptional priming is effective 10–30 min after TLR4 and TLR2 binding and depends on the MyD88/IL-1 receptor-associated kinase 1/4 (IRAK1/4) pathway. Notably, IRAK1, which is required for this process and associates with NLRP3–ASC complexes, is degraded after 30 min providing a possible mechanism for the transient nature of this priming.Citation44 Non-transcriptional priming is also effective 30 min–1 h after TLR4 and TLR3 binding and then depends on the activation of the TRIF/IRAK1/FADD/caspase-8/receptor-interacting protein 1 (RIP1) pathway.Citation37,Citation42–Citation45 These pathways are referred to as early and intermediate priming, respectively, and both rely on mitochondrial ROS (mtROS), which can even bypass the requirement for TLR agonists.Citation42 In addition, a third non-transcriptional priming pathway has been described in macrophages deficient for caspase-8 activity. This priming pathway would bypass the requirement for NLRP3 activation and rely on FADD and mixed lineage kinase domain like (MLKL) pseudokinase.Citation38 However, ongoing necroptosis in this setting is likely to provide NLRP3 activation signal.Citation37,Citation45,Citation46
Finally, prolonged exposure (>12 h) of macrophages to TLR4 agonists, but not TNF or TLR2/1 agonists, renders NLRP3 refractory to further activation signals. The requirement for coordinately received priming and activation signals may physiologically limit inappropriate response, and this desensitization process may therefore protect the organism against detrimental inflammation. Several mechanisms support NLRP3 desensitization. LPS triggers IFN-I secretion which induces, in an autocrine/paracrine manner, the expression of inducible nitric oxide synthase (iNOS) and TRIM30.Citation47,Citation48 iNOS inhibits NLRP3 through the production of endogenous NO,Citation47 while TRIM30 inhibits NLRP3 by reducing the production of ROS.Citation48 In addition, IFN-I also represses the expression of the inflammasome substrates pro-IL-1a/β through the secretion of IL-10.Citation49 Moreover, LPS-inducible A20 dampens pro-IL-1β maturation by caspase-1 (detailed in the “Post-translational modifications of ASC, caspase-1, and IL-1β in the regulation of the inflammasome” section).Citation50 Finally, the induction of Sequestosome 1 (SQSTM1, alternatively named p62) and degradation of damaged mitochondria that emit inflammasome-activating signals also participate in this desensitization.Citation51 On the contrary, prolonged exposure to TNF does not make NLRP3 refractory to further activation, and may thus participate in NLRP3 priming involved in chronic inflammatory diseases, such as CAPS and Alzheimer’s disease. Indeed, β-amyloid fibrils as well as inflammation associated with aging induce the release of TNF.Citation33,Citation36,Citation52,Citation53
In summary, successive signaling pathways activated downstream of PRRs, cytokine receptors, and other priming receptors control one or several of the following events: 1) the early or intermediate switch in NLRP3 by post-translational modifications to a form competent for further activation; 2) the late transcriptional upregulation of NLRP3, inflammasome substrates including pro-IL-1β, and probably other key regulator(s); and 3) upon their prolonged activation, the inhibition of subsequent inflammasome activation by targeting NLRP3, inflammasome substrate pro-IL-1β, or key elements in NLRP3 activation pathways such as ROS or mitochondria. The impact and the relevance of these successive signaling pathways in infectious or auto-inflammatory contexts in vivo remain poorly understood.
NLRP3 inflammasome activation
Priming licenses NLRP3 to subsequent activation by highly diverse agents and conditions. These activation signals can be classified according to the type of cellular stress that they cause (). Agents leading to changes in the plasma membrane permeability to ions form the first category. It comprises, for example, pore-forming bacterial toxins including nigericin,Citation54 as well as the membrane attack complex assembled upon the activation of the complement system.Citation55,Citation56 As already mentioned, permeabilization of plasma membrane by GSDMD pores is critical in NLRP3 activation downstream of non-canonical inflammasomes.Citation13,Citation17 Finally, extracellular ATP, as released by necrotic neighboring cells, activates NLRP3 through the plasma membrane P2X7 receptor and the subsequent opening of ionic pores.Citation57 Crystals constitute a second class of NLRP3 activation signals including asbestos, silica, cholesterol crystals, β-amyloid, and liposomes.Citation58–Citation61 They are phagocytized but resistant to degradation, and their accumulation ultimately leads to the rupture of phagolysosomes and the consequent release of cathepsins and Ca2+ in the cytosol, and both may account for inflammasome activation.Citation58,Citation62 Finally, as mentioned before, particulates formed by inflammasome specks released in the extracellular milieu by pyroptotic cells may play a key role in inflammation amplification.Citation15,Citation16 Their phagocytosis by neighboring macrophages leads to NLRP3 activation by lysosomal disruption similar to other particulate signals, and in addition triggers direct recruitment and activation of cellular caspase-1 by these ASC specks. In addition, extracellular ASC speck has been recently shown to recruit β-amyloid and seed aggregates, spreading β-amyloid pathology in Alzheimer’s disease.Citation63 Agents and conditions triggering mitochondrial dysfunction constitute the third class of NLRP3 activators, including metabolic stress, inhibitors of respiratory chain complexes, and blockade of mitophagy.Citation64 Finally, other NLRP3 activation signals cannot be included in any of these classes as, for example, ultraviolet radiations and activation of G-protein-coupled receptors by the extracellular Ca2+.Citation62,Citation65–Citation67 These highly diverse activation signals seem to engage several pathways to activate NLRP3.Citation68
Figure 4 NLRP3 activation.
Notes: NLRP3 activation is triggered by various stress signals including: 1) increase in plasma membrane permeability caused by bacterial toxins, activation of P2X7R by ATP release from necrotic cells, complement membrane attack complex (MAC) or GSDMD pore formed downstream of the activation of the non-canonical inflammasome by cytosolic LPS, 2) lysosome rupture caused by phagocytosis of particulates including crystals (asbestos, silica, monosodium urate [MSU], cholesterol crystals, etc) as well as ASC specks released from neighboring cells, and 3) mitochondrial damages. NLRP3 activation is highly dependent on K+ efflux. In addition, increase in membrane permeability, lysosome rupture, and activation of G-protein coupled receptor (GPCR) lead to a rise in cytosolic Ca2+. Subsequent Ca2+ overload induces mitochondrial damage, then cardiolipin exposure and oxidized mtDNA participate in NLRP3 activation at mitochondria-associated ER membranes (MAMs), resulting in the inflammasome assembly and caspase-1 activation. In addition, exogenous ASC specks released in the cytosol following lysosome rupture can directly activate caspase-1 and therefore propagate rapidly the inflammatory signal to neighboring cells.
Abbreviations: TLR, Toll-like receptor; GSDMD, gasdermin-D; LPS, lipopolysaccharide; ASC, apoptosis-associated speck-like protein containing a CARD domain; mtDNA, mitochondrial DNA; ER, endoplasmic reticulum.
![Figure 4 NLRP3 activation.Notes: NLRP3 activation is triggered by various stress signals including: 1) increase in plasma membrane permeability caused by bacterial toxins, activation of P2X7R by ATP release from necrotic cells, complement membrane attack complex (MAC) or GSDMD pore formed downstream of the activation of the non-canonical inflammasome by cytosolic LPS, 2) lysosome rupture caused by phagocytosis of particulates including crystals (asbestos, silica, monosodium urate [MSU], cholesterol crystals, etc) as well as ASC specks released from neighboring cells, and 3) mitochondrial damages. NLRP3 activation is highly dependent on K+ efflux. In addition, increase in membrane permeability, lysosome rupture, and activation of G-protein coupled receptor (GPCR) lead to a rise in cytosolic Ca2+. Subsequent Ca2+ overload induces mitochondrial damage, then cardiolipin exposure and oxidized mtDNA participate in NLRP3 activation at mitochondria-associated ER membranes (MAMs), resulting in the inflammasome assembly and caspase-1 activation. In addition, exogenous ASC specks released in the cytosol following lysosome rupture can directly activate caspase-1 and therefore propagate rapidly the inflammatory signal to neighboring cells.Abbreviations: TLR, Toll-like receptor; GSDMD, gasdermin-D; LPS, lipopolysaccharide; ASC, apoptosis-associated speck-like protein containing a CARD domain; mtDNA, mitochondrial DNA; ER, endoplasmic reticulum.](/cms/asset/7b8f9ee3-64ca-4667-b2dd-c2bbc4a45560/djir_a_141220_f0004_c.jpg)
Although a unifying model for NLRP3 activation still remains controversial, common key signaling features observed downstream of several activators recently emerged including in particular changes in cytosolic ionic concentration and mitochondrial damage. A decrease in cytosolic K+ is observed upon exposure to extracellular ATP, pore-forming toxins, and particulates.Citation69 K+ efflux precedes mitochondrial dysfunctions, and correlates with inflammasome activation upon experimental up- or down-modulations of extracellular K+ concentrations, suggesting a key role of reduction in cytosolic K+ levels in the upstream control of NLRP3 activation. The mechanism of this control remains unknown, although it has been suggested that low cytosolic K+ concentration might participate directly or indirectly in the conformational changes of NLRP3 oligomers associated with inflammasome formation.Citation6 Rise in cytosolic Ca2+ is also associated with inflammasome activation following numerous stimuli.Citation62 High extracellular K+ reduces cytosolic Ca2+ rise, suggesting that K+ efflux may promote Ca2+ influx to activate NLRP3, although its significance remains debated since the increase in cytosolic Ca2+ is not sufficient to trigger NLRP3 activation and its role may also be restricted to K+ efflux counterbalancing.Citation69 Several ionic channels including transient receptor potential, inositol tri-phosphate receptor, and ryanodine receptor have been reported to mediate Ca2+ fluxes associated to NLRP3 activation depending on the inflammatory stimuli and contexts.Citation6,Citation56,Citation61,Citation62,Citation65 In addition, disruption of lysosomes following particulate phagocytosis may lead to the release of lysosomal Ca2+ into the cytosol and may participate in the rise of cytosolic Ca2+ levels.Citation62 Increased Ca2+ concentration favors ASC recruitment to NLRP3.Citation65 Lastly, mitochondrial damage constitutes another shared feature of NLRP3 activation pathways. This mitochondrial dysfunction can be caused after mitochondrial Ca2+ overload following rise in cytosolic Ca2+ concentration.Citation62 The events following mitochondrial damage such as loss of membrane potential (∆Ψm), production of ROS, release of mitochondrial DNA (mtDNA) in the cytosol, and exposure of cardiolipin at the outer membrane of mitochondria have all been proposed to mediate NLRP3 activation. In particular, mtDNA and cardiolipin could be direct ligands of NLRP3.Citation70–Citation72 Accordingly, inhibition of mitophagy promotes NLRP3 activation through the accumulation of defective mitochondria.Citation72
NLRP3 inflammasome assembly is similarly controlled in most of other innate immune cells, including murine monocytes, mast cells, and neutrophils.Citation73–Citation75 Noteworthy, in human monocytes, strong LPS priming signals have been reported to be sufficient to trigger NLRP3 inflammasome activation in some conditions, although activation signals remain necessary for potentiation of weak priming signals.Citation73,Citation76–Citation78 These differential requirements may be due to ATP secretion by human monocytes upon priming. Released ATP may then act as an activation signal in an autocrine/paracrine manner.Citation76,Citation77 More recently, it has been proposed that in human monocytes, an alternative inflammasome pathway may be triggered without the requirement of activation signals.Citation73 This species-specific alternative pathway is independent of K+ efflux and leads to a low and gradual IL-1β response without pyroptosome formation and therefore significantly differs from the all-or-nothing speck response triggered by canonical activation stimuli. This alternative pathway depends on TRIF, RIPK1, FADD, and caspase-8 signaling pathways as evidenced in BLaER1 post-mitotic monocytes transdifferentiated from immortalized B cells.
NLRP3 trafficking toward inflammasome assembly
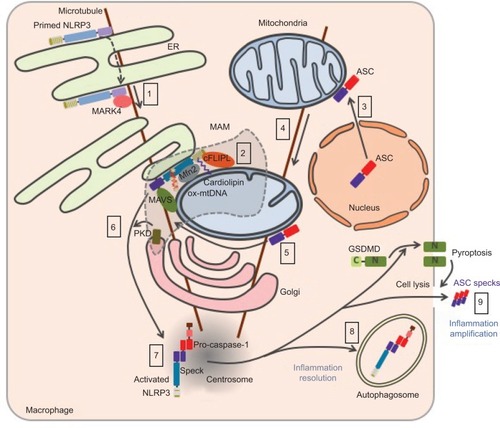
The trafficking of NLRP3 seems to be tightly regulated upon both priming and activation processes and appears to be critical for inflammasome assembly (). Nevertheless, the current model remains controversial due to possible artifacts of overexpression approaches, concerns on the specificity of anti-NLRP3 antibodies raising doubts on endogenous staining, and extensive use of artificial cell lines. NLRP3 is both cytosolic and associated with the ER in THP1 and resting bone marrow-derived macrophages (BMDMs).Citation64,Citation79 Upon NLRP3 activation, NLRP3 is recruited to mitochondria-associated ER membranes (MAMs).Citation64,Citation71,Citation80 This recruitment is independent of ASC and therefore probably precedes NLRP3 interaction with its adaptor.Citation80,Citation81 Consistently, NLRP3 recruitment to the mitochondria has been recently reported to occur during priming.Citation82 Consistent with the inhibition of NLRP3 pathway by colchicine or nocodazole,Citation79 NLRP3 co-migrates with LPS-induced microtubule-affinity regulating kinase 4 (MARK4) along microtubules to reach mitochondria, suggesting a role of microtubules in NLRP3 recruitment to the MAMs.Citation83 MARK4 kinase domain interacts with NLRP3 PYD and NACHT domains, and its expression is required for NLRP3 localization to mitochondria. Other interactors of NLRP3 have been described to be required for – or to facilitate – NLRP3 relocation and activation including cardiolipin, normally located in the inner mitochondrial membrane, that interacts with NLRP3 LRR domain,Citation71 mitochondrial antiviral signaling (MAVS) that interacts with NLRP3 PYD,Citation80 cellular FLICE-inhibitory protein long form (cFLIPL) upregulated by TLR signaling and interacting with NLRP3 LRR domain.Citation84,Citation85 Noteworthy, the role of MAVS remains controversial and has been differentially shown by others to be restricted to NLRP3 control downstream of RNA virus infections, and implicated in the priming of NLRP3 and ASC stabilization.Citation86,Citation87 In addition, ∆Ψm-dependent binding of NLRP3 with mitofusin-2 (Mfn 2) may potentiate its interaction with MAVS, upon RNA virus infections, but the role of mitofusin-2 in NLRP3 recruitment to the mitochondria has not been investigated.Citation88 Finally, small heterodimer partner (SHP) that co-translocates to the mitochondria upon NLRP3 activation and interacts with its PYD domain acts as a negative regulator. Interestingly, SHP deficiency abolishes NLRP3 recruitment to the mitochondria suggesting a not yet characterized role in NLRP3 trafficking.Citation81 Other inflammasome components have also been shown to relocate upon priming or activation. While caspase-1 is cytosolic in basal condition, it is recruited to the mitochondria upon priming probably through its interaction with externalized cardiolipin.Citation82 In basal condition, ASC has been reported to be mostly in the nucleus but is also found in the cytoplasm where it associates with mitochondria.Citation79,Citation89 Upon NLRP3 activation, its mitochondrial recruitment increases.Citation82 In addition, mitochondria then relocate in a microtubule network-dependent manner to approach the ER at the perinuclear region where acetylated tubulin accumulates.Citation79 Inhibition of a-tubulin deacetylase sirtuin 2 (SIRT2) caused by mitochondrial damage and subsequent decrease in NAD+ may induce this process.Citation79 NLRP3 undergoes self-oligomerization and interacts with ASC in the MAMs before being released in the cytosol.Citation81,Citation90 Clustering of mitochondria around the Golgi and phosphorylation of NLRP3 by Golgi-associated protein kinase D (PKD, detailed hereunder) control this release.Citation90 NLRP3 and ASC then colocalize in one cytosolic perinuclear speck of 1–6 µm.Citation89,Citation91 This speck has recently been described to localize at the centrosome, suggesting a role of microtubule network in speck formation and providing a mechanistic explanation for the formation of a single speck per cell.Citation83 Consistently, the centrosomal protein NIMA-related protein kinase 7 (NEK7) associates with the inflammasome complex.Citation92–Citation94 On the contrary, a previous study reported the speck to be distinct from the centrosome.Citation95 Finally, inflammasome targeting to autophagosome plays a role in the control or resolving of the response.Citation96,Citation97 If the inflammasome is not degraded by autophagosome, the abovementioned release of specks participates in the amplification of the inflammatory response.Citation15,Citation16
Figure 5 Current model of NLRP3 subcellular localization during its activation process.
Notes: Primed NLRP3 is both cytosolic and associated with endoplasmic reticulum (eR). Upon activation, NLRP3 is recruited to mitochondria-associated ER membranes (MAMs) by co-migration with MARK4 along microtubules (1). NLRP3 is stabilized in the MAMs through its interaction with cardiolipin, MAVS, cFLIPL, Mfn 2, and oxidized mtDNA at the surface of the mitochondria (2). Upon NLRP3 activation, ASC relocates from the nucleus to the mitochondria network (3), which approaches the ER in a microtubule-dependent manner (4). NLRP3 associates with ASC in the MAMs (5). Phosphorylation of NLRP3 by Golgi-associated PKD leads to the release of NLRP3/ASC complex from the MAMs (6) and assembly of a single speck at the centrosome (7). Autophagy dampens inflammation by targeting the inflammasome (8), while release of ASC speck from pyroptotic cells propagates the inflammatory signal to neighboring cells (9).
Abbreviations: MAVS, mitochondrial antiviral signaling; mtDNA, mitochondrial DNA; ASC, apoptosis-associated speck-like protein containing a CARD domain; PKD, protein kinase D; mtDNA, mitochondrial DNA; GSDMD, gasdermin-D.
To conclude, the assembly of the NLRP3 inflammasome in macrophages is a tightly controlled event that requires coordinated priming and activation signals. Signaling pathways and molecular mechanisms involved are not yet fully understood. Notably, accumulative studies recently highlight the role of NLRP3 post-translational modifications in both processes, that is, priming and activation (; ).
Table 1 Post-translational modifications of NLRP3 inflammasome
Figure 6 Post-translational modifications of NLRP3 involved in its priming, activation, and desensitization.
Notes: Stimuli, enzymes, modifications, and targeted residues that promote or inhibit inflammasome assembly are represented in pink and green, respectively.
Abbreviations: LPS, lipopolysaccharide; ASC, apoptosis-associated speck-like protein containing a CARD domain; Nig, nigericin; Sil, silica; MSU, monosodium urate; PYD, pyrin domain.
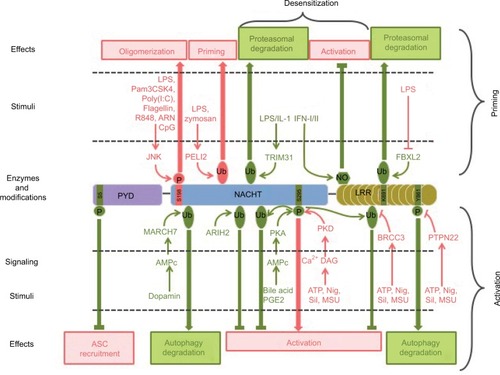
NLRP3 post-translational modifications as a regulation mechanism of inflammasome assembly
NLRP3 post-translational modifications in inflammasome priming
As already mentioned, NLRP3 priming can occur independently of new protein synthesis, and therefore relies primarily on post-translational modifications in addition to the extra need of transcriptional gene upregulation in some conditions.Citation42 Phosphorylation of S198 (corresponding to S194 in mouse NLRP3) by JNK1 has been recently proposed as a universal feature of NLRP3 priming, required for both transcriptional-dependent and -independent priming pathways, and downstream of both MyD88 and TRIF signaling.Citation98 S198 phosphorylation kinetics correlates with NLRP3 priming by these different pathways. S198 phosphorylation leads to NLRP3 deubiquitination and promotes its oligomerization. S198N polymorphism has been identified in CAPS patients.Citation99 This observation is surprising since CAPS is typically caused by gain of function mutations in NLRP3. Nevertheless, this patient bore another well characterized CAPS mutation Y570C that was probably causing over-activation of NLRP3 without the need of S198 phosphorylation. These results are consistent with earlier findings that NLRP3 is constitutively ubiquitinated and that non-transcriptional priming induced by short LPS exposure leads to NLRP3 deubiquitination in a mtROS-dependent manner.Citation42 More recently and in opposite to its constitutive ubiquitination model, NLRP3 has been reported to be very transiently ubiquitinated with K63 chains following LPS priming.Citation100 This ubiquitination depends on the E3 ligase Pellino 2, whose deficiency significantly impairs NLRP3 response in cultured BMDMs, as well as in several mouse models. Most E3 ubiquitin ligases and deubiquitinases controlling NLRP3 priming remain to be identified. Noteworthy, chaperones such as suppressor of the G2 allele of skp1 (SGT1) and heat shock protein 90 (HSP90), which have E3 ligase activity, associate with NLRP3 in resting conditions.Citation101 In addition, the increase in NLRP3 level upon transcriptional priming may in part rely on stabilization of NLRP3 through post-translational regulation. Indeed, NLRP3 ubiquitination on K691 (K689 in mouse) by skp-cullin-F box L2 (SCFFBXL2) E3 ligase targets NLRP3 to proteasomal degradation.Citation102,Citation103 Transcriptional priming leads to the expression of the SCFFBXO3 E3 ligase that destabilizes SCFFBXL2 via ubiquitination and degradation, resulting in increased NLRP3 half-life. Other NLRP3 modifications have been reported upon LPS priming including S-glutathionylation, but their relevance in the priming process remains to be established.Citation104
NLRP3 post-translational modifications in inflammasome activation
Accumulative studies point to the critical role of NLRP3 post-translational modifications in NLRP3 activation step. We first described that NLRP3 inflammasome assembly requires NLRP3 deubiquitination by the BRCA1/BRCA2-containing complex subunit 3 (BRCC3) deubiquitinase downstream of all tested activation signals.Citation105 Therefore, this deubiquitination can be considered as a key common regulatory mechanism of inflammasome activation. It does not depend on mtROS, and therefore signaling differs from deubiquitination associated with NLRP3 priming.Citation42 NACHT and LRR domains of NLRP3 are independently ubiquitinated, and both K48 and K63 ubiquitin chains associate with NLRP3.Citation105 The K63-specific deubiquitinase BRCC3 specifically targets the NLRP3 LRR domain, but the precise ubiquitinated residue and the structural consequences of this deubiquitination remain unknown. BRCC3 inhibition promotes NLRP3 ubiquitination and blocks inflammasome assembly but does not target NLRP3 for degradation.Citation105 Consistently, the ARIH2 ubiquitin ligase has recently been identified in the speck.Citation106 While ARIH2 promotes NLRP3 ubiquitination with K63 and K48 chains and inhibits the inflammasome activation, it does not reduce NLRP3 stability. In addition, NLRP3 activation is also downregulated by degradative ubiquitination. The inflammasome inhibitor dopamine induces an increase in cAMP level through dopamine receptor D1 (DRD1).Citation107 cAMP then binds to NLRP3 and triggers the ubiquitination of NLRP3 NACHT and LRR domains with K48 ubiquitin chains by the E3 ubiquitin ligase membrane associated ring-CH-type finger 7 (MARCH7), targeting NLRP3 to autophagy-mediated degradation.Citation107
Phosphorylation of NLRP3 also controls its activation process. Phosphorylation of S295 (S291 in mouse NLRP3) in the NACHT domain has recently emerged as a key regulatory point in NLRP3 activation process. Initially, bile acids binding Takeda G-protein-coupled receptor 5 (TGR5) and eicosanoid prostaglandin E2 (PGE2) binding E prostanoid 4 receptor (EP4) have been shown to inhibit NLRP3 through production of cAMP, activation of protein kinase A (PKA), phosphorylation of NLRP3 by PKA on S295 leading to its inactivation by non-degradative ubiquitination.Citation108,Citation109 On the opposite, S295 phosphorylation by PKDs has recently been reported to be required for NLRP3 activation.Citation90 Indeed, in response to inflammasome activators, Golgi-associated diacyl-glycerol (DAG) recruits PKDs while mitochondria cluster to the Golgi. Then PKDs phosphorylate MAM-associated NLRP3 on S295, leading to its release from the MAMs and inflammasome assembly in the cytosol. PKDs are dispensable for mitochondrial clustering to the Golgi and act downstream of mitochondrial damage. Reconciling all data, both NLRP3 phosphonull and phospho-mimetic mutants are inactive, suggesting that S295 phosphorylation needs to be timely and locally regulated. Indeed, S295 phosphorylation initially prevents NLRP3 recruitment to MAMs necessary for its activation, while S295 phosphorylation later by PKDs allows its release from the MAMs to form cytosolic specks.Citation90 In addition, inflammasome assembly also requires the dephosphorylation of S5 (S3 in mouse NLRP3) in the PYD of NLRP3.Citation110 Indeed, phosphorylation of S5 prevents ASC recruitment by electrostatic repulsion. Protein phosphatase 2A (PP2A) could be involved in this dephosphorylation.Citation110,Citation111 However, it remains unclear whether S5 dephosphorylation occurs during the priming or the activation step. Noteworthy, the critical role of kinase NEK7 in NLRP3 inflammasome assembly has recently been discovered by several independent studies.Citation92–Citation94 NEK7 interacts with NLRP3 LRR domain and takes part in the inflammasome complex. However, its catalytic activity is dispensable for the inflammasome activity and therefore does not regulate inflammasome assembly by direct phosphorylation of NLRP3 or other components of the complex but rather by scaffolding, subcellular relocation, or other functions that remain to be elucidated. Finally, the phosphorylation on Y861 (Y859 in mouse) in the LRR domain dampens the inflammasome activity by targeting NLRP3 to autophagosomes.Citation97,Citation112 Phosphorylated NLRP3 interacts with SQSTM1 in an ASC-dependent manner, suggesting that this downregulation may occur later in the process after inflammasome assembly.Citation97 Upon activation signals, protein tyrosine phosphatase non-receptor type 22 (PTPN22) is recruited to NLRP3 in an ASC-dependent manner and sustains IL-1β secretion by dephosphorylating Y861.Citation112 NLRP3 dephosphorylation in a model of dextran sulfate sodium-induced colitis requires PTPN22 and correlates with caspase-1, IL-1β, and IL-18 maturation as well as granulocyte recruitment, suggesting that NLRP3 Y861 phosphorylation may play a significant role in inflammatory diseases. Noteworthy, PTPN22 is genetically associated with several inflammatory disorders linked to inflammasome dysregulation.
NLRP3 post-translational modifications involved in its desensitization upon prolonged priming
As mentioned previously, prolonged TLR4-mediated priming in the absence of timely activation causes NLRP3 to be refractory to late activation stimuli. This desensitization probably participates in maintaining NLRP3 under tight control preventing inappropriate inflammasome-mediated inflammation and relies partly on NLRP3 post-translational modifications. Indeed, the E3 ubiquitin ligase TRIM31, which is induced after LPS and IL-1β, prolonged priming triggers K48 ubiquitination of NLRP3 and leads to its proteasomal degradation.Citation113 In addition, as already mentioned, Y861 phosphorylation targets the inflammasome to autophagosomes preventing NLRP3 activation.Citation97,Citation112 Therefore, this mechanism may support desensitization upon prolonged priming. Last but not least, independent studies evidenced the key role of iNOS in this desensitization of NLRP3.Citation47,Citation114 Prolonged exposure to LPS triggers IFN-I secretion, which in turn induces iNOS expression in an autocrine/paracrine manner. iNOS inhibits NLRP3 at least in part through its direct modifications. Indeed, the subsequent production of NO leads to the S-nitrosylation of the cysteine-rich LRR domain of NLRP3, a modification that prevents ASC oligomer formation.Citation47 Notably, IFN-β inhibits NLRP3 through the same mechanism.Citation115
Post-translational modifications of ASC, caspase-1, and IL-1b in the regulation of the inflammasome
The NLRP3 inflammasome activity is also regulated by post-translational modifications of its other components including ASC, caspase-1, and its substrate IL-1β. ASC linear ubiquitination by hemeoxidized IRP2 ubiquitin ligase 1L (HOIL-1L) in the linear ubiquitin chain assembly complex (LUBAC) is required for its recruitment to NLRP3, inflammasome assembly, and activation.Citation116 However, whether ASC linear ubiquitination occurs downstream of priming or activation signaling has not yet been investigated. Besides, ubiquitination of ASC at K174 residue with K63 chains by tumor necrosis factor receptor-associated factor 3 (TRAF3) downstream of MAVS stabilizes ASC and supports inflammasome assembly upon RNA virus infections.Citation87 Interestingly, ASC is also ubiquitinated with K63 chains upon LPS priming, and later deubiquitinated by ubiquitin specific peptidase 50 (USP50) upon NLRP3 activation, suggesting a conserved regulation of speck formation by dynamic ASC K63 ubiquitination.Citation117 Finally, ASC ubiquitination with K63 ubiquitin chain also participates in the resolution of the response by targeting the inflammasome to degradation through the autophagy machinery.Citation96 Phosphorylation of ASC also likely plays a role in inflammasome regulation. A previous report shows that ectopically expressed ASC PYD domain can be phosphorylated in response to TNF treatment.Citation118 More recently, ASC has been shown to be phosphorylated upon inflammasome activation stimuli through an unknown pathway dependent on both spleen tyrosine kinase (SYK) and c-Jun N-terminal kinase (JNK), which are required for ASC oligomerization and speck formation but not for ASC recruitment to NLRP3. Among several other phosphorylated residues, Y146 in ASC CARD domain (Y144 in mouse ASC) seems to be the critical target of this pathway.Citation119 The inflammasome effector caspase-1 has been reported to be ubiquitinated in its CARD domain with K63 ubiquitin chains by cellular inhibitor of apoptosis 2 (cIAP-2) upon NLRP3 activation signals.Citation120 Since cIAP-1/2 and TRAF2-containing complex, which associates with caspase-1, is required for IL-1β secretion, this non-degradative ubiquitination may promote inflammasome activity but the mechanism remains unknown. In addition, caspase-1 is also unexpectedly directly inhibited by some ROS. Indeed, accumulation of intracellular superoxide, as observed in superoxide dismutase 1 (SOD-1) deficiency, leads to caspase-1 S-glutathionylation on C362 and C397 and its inhibition.Citation121 Finally, post-translational modifications of the caspase-1 substrate pro-IL-1β also impact inflammasome activity. Indeed, pro-IL-1β associates with K63 ubiquitin chains on K133 and unanchored K48 chains in a complex pre-formed upon LPS transcriptional priming and containing RIPK1/3, pro-caspase-1/8, and A20.Citation50 While A20 reduces pro-IL-1β K63 ubiquitination and maturation, on the contrary RIPK3 increases pro-IL-1β association with ubiquitin chains and promotes its spontaneous NLRP3-dependent maturation upon A20 deficiency. Therefore, pro-IL-1β ubiquitination supports its caspase-1-mediated proteolytic processing. It may be interesting in future studies to address ubiquitination of other caspase-1 substrates including pro-IL-18 and GSDMD.
Conclusion
While inflammation is important for organism protection, its dysregulation is highly detrimental as tragically exemplified by a number of inflammatory diseases. Accordingly, NLRP3 inflammasome assembly is strictly regulated by the requirement of coordinated priming and activation signals. These signals are highly diverse, and despite almost 20 years of research, our understanding of the signaling pathways and the molecular mechanisms controlling NLRP3 remains largely incomplete. Post-translational modifications of NLRP3 and other inflammasome components have recently emerged as an important mechanism in the control of its assembly. In the near future, ongoing characterization of the molecular consequences of these modifications will fill some of these gaps. In addition, identification of the enzymes and their co-factors controlling these modifications may lead to the identification of novel druggable targets for specific anti-inflammatory therapies.
Acknowledgments
This work was supported in part by grants to BFP from the Agence Nationale pour la Recherche (ANR-13-JSV3-0002-01) and the European Research Council (ERC-2013-CoG_616986). We thank Pablo Palazon-Riquelme, Danish Patoli, and Amaury Rey for comments on the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
- MedzhitovROrigin and physiological roles of inflammationNature2008454720342843518650913
- MedzhitovRJanewayCAInnate immunity: the virtues of a nonclonal system of recognitionCell19979132952989363937
- MatzingerPTolerance, danger, and the extended familyAnnu Rev Immunol19941299110458011301
- HuZZhouQZhangCStructural and biochemical basis for induced self-propagation of NLRC4Science2015350625939940426449475
- Fernandes-AlnemriTYuJ-WDattaPWuJAlnemriESAIM2 activates the inflammasome and cell death in response to cytoplasmic DNANature2009458723750951319158676
- CompanVBaroja-MazoALópez-CastejónGCell volume regulation modulates NLRP3 inflammasome activationImmunity201237348750022981536
- LuAMagupalliVGRuanJUnified polymerization mechanism for the assembly of ASC-dependent inflammasomesCell201415661193120624630722
- MasumotoJTaniguchiSAyukawaKASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cellsJ Biol Chem199927448338353383810567338
- DickMSSborgiLRühlSHillerSBrozPASC filament formation serves as a signal amplification mechanism for inflammasomesNat Commun201671192927329339
- ShiJZhaoYWangYInflammatory caspases are innate immune receptors for intracellular LPSNature2014514752118719225119034
- SchroderKTschoppJThe inflammasomesCell2010140682183220303873
- KayagakiNStoweIBLeeBLCaspase-11 cleaves gas-dermin D for non-canonical inflammasome signallingNature2015526757566667126375259
- AgliettiRAEstevezAGuptaAGsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranesProc Natl Acad Sci U S A2016113287858786327339137
- BergsbakenTFinkSLCooksonBTPyroptosis: host cell death and inflammationNat Rev Microbiol2009729910919148178
- Baroja-MazoAMartín-SánchezFGomezAIThe NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory responseNat Immunol201415873874824952504
- FranklinBSBossallerLDe NardoDThe adaptor ASC has extracellular and “prionoid” activities that propagate inflammationNat Immunol201415872773724952505
- RühlSBrozPCaspase-11 activates a canonical NLRP3 inflammasome by promoting K+ effluxEur J Immunol201545102927293626173909
- HoffmanDHMFamilial cold autoinflammatory syndromeJ Allergy Clin Immunol2001108461562011590390
- ShinkaiKMccalmontTHLeslieKSCryopyrin-associated periodic syndromes and autoinflammationClin Exp Dermatol20083311917927785
- MartinonFPétrilliVMayorATardivelATschoppJGout-associated uric acid crystals activate the NALP3 inflammasomeNature2006440708123724116407889
- VandanmagsarBYoumY-HRavussinAThe NALP3/NLRP3 inflammasome instigates obesity-induced autoinflammation and insulin resistanceNat Med201117217918821217695
- YoumY-HGrantRWMcCabeLRCanonical Nlrp3 inflamma-some links systemic low grade inflammation to functional decline in agingCell Metab201318451953224093676
- HenekaMTKummerMPLatzEInnate immune activation in neurodegenerative diseaseNat Rev Immunol201414746347724962261
- FranchiLMuñoz-PlanilloRNúñezGSensing and reacting to microbes via the inflammasomesNat Immunol201213432533222430785
- JoE-KKimJKShinD-MSasakawaCMolecular mechanisms regulating NLRP3 inflammasome activationCell Mol Immunol201613214815926549800
- RatsimandresyRADorfleutnerAStehlikCAn update on PYRIN domain-containing pattern recognition receptors: from immunity to pathologyFront Immunol2013444024367371
- ElliottEISutterwalaFSMonocytes take their own path to IL-1βImmunity201644471371527096310
- AlexiouPChatzopoulouMPegklidouKDemopoulosVJRAGE: a multi-ligand receptor unveiling novel insights in health and diseaseCurr Med Chem201017212232225220459381
- XiangMShiXLiYHemorrhagic shock activation of NLRP3 inflammasome in lung endothelial cellsJ Immunol201118794809481721940680
- FritzJHFerreroRLPhilpottDJGirardinSENod-like proteins in immunity, inflammation and diseaseNat Immunol20067121250125717110941
- MeylanETschoppJKarinMIntracellular pattern recognition receptors in the host responseNature20064427098394416823444
- MitchellJAPaul-ClarkMJClarkeGWMcMasterSKCartwrightNCritical role of toll-like receptors and nucleotide oligomerisation domain in the regulation of health and diseaseJ Endocrinol2007193332333017535871
- FranchiLEigenbrodTNúñezGCutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulationJ Immunol2009183279279619542372
- SutterwalaFSHaaskenSCasselSLMechanism of NLRP3 inflammasome activationAnn N Y Acad Sci20141319829524840700
- BauernfeindFGHorvathGStutzACutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expressionJ Immunol2009183278779119570822
- BauernfeindFNiepmannSKnollePAHornungVAging-associated TNF production primes inflammasome activation and NLRP3-related metabolic disturbancesJ Immunol Baltim Md 19502016197729002908
- GurungPAnandPKMalireddiRKSFADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomesJ Immunol201419241835184624453255
- ZhangXFanCZhangHMLKL and FADD are critical for suppressing progressive lymphoproliferative disease and activating the NLRP3 inflammasomeCell Rep201616123247325927498868
- BauernfeindFBartokERiegerAFranchiLNúñezGHornungVCutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasomeJ Immunol Baltim Md 195020111872613617
- JulianaCFernandes-AlnemriTWuJAnti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasomeJ Biol Chem2010285139792980220093358
- SchroderKSagulenkoVZamoshnikovaAAcute lipopolysaccha-ride priming boosts inflammasome activation independently of inflammasome sensor inductionImmunobiology2012217121325132922898390
- JulianaCFernandes-AlnemriTKangSFariasAQinFAlnemriESNon-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activationJ Biol Chem201228743366173662222948162
- Fernandes-AlnemriTKangSAndersonCSagaraJFitzgeraldKAAlnemriESCutting edge: TLR signaling licenses IRAK1 for rapid activation of the NLRP3 inflammasomeJ Immunol201319183995399924043892
- LinK-MHuWTroutmanTDIRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activationProc Natl Acad Sci U S A2014111277578024379360
- KangSFernandes-AlnemriTRogersCCaspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3Nat Commun20156751526104484
- KangT-BYangS-HTothBKovalenkoAWallachDCaspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasomeImmunity2013381274023260196
- Hernandez-CuellarETsuchiyaKHaraHCutting edge: nitric oxide inhibits the NLRP3 inflammasomeJ Immunol2012189115113511723100513
- HuYMaoKZengYTripartite-motif protein 30 negatively regulates NLRP3 inflammasome activation by modulating reactive oxygen species productionJ Immunol Baltim Md 195020101851276997705
- GuardaGBraunMStaehliFType I interferon inhibits interleukin-1 production and inflammasome activationImmunity201134221322321349431
- DuongBHOnizawaMOses-PrietoJAA20 restricts ubiquitination of pro-interleukin-1β protein complexes and suppresses NLRP3 inflammasome activityImmunity2015421556725607459
- ZhongZUmemuraASanchez-LopezENF-κB restricts inflammasome activation via elimination of damaged mitochondriaCell2016164589691026919428
- McGeoughMDWreeAInzaugaratMETNF regulates transcription of NLRP3 inflammasome components and inflammatory molecules in cryopyrinopathiesJ Clin Invest2017127124488449729130929
- CasalCSerratosaJTusellJMRelationship between beta-AP peptide aggregation and microglial activationBrain Res20029281–2768411844474
- CooksonBTBrennanMAPro-inflammatory programmed cell deathTrends Microbiol20019311311411303500
- LaudisiFSpreaficoREvrardMCutting edge: the NLRP3 inflammasome links complement-mediated inflammation and IL-1β releaseJ Immunol201319131006101023817414
- TriantafilouKHughesTRTriantafilouMMorganBPThe complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activationJ Cell Sci2013126132903291323613465
- SolleMLabasiJPerregauxDGAltered cytokine production in mice lacking P2X(7) receptorsJ Biol Chem2001276112513211016935
- HornungVBauernfeindFHalleASilica crystals and aluminum salts mediate NALP-3 inflammasome activation via phagosomal destabilizationNat Immunol20089884785618604214
- DuewellPKonoHRaynerKJNLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystalsNature201046472931357136120428172
- HalleAHornungVPetzoldGCThe NALP3 inflammasome is involved in the innate immune response to amyloid-betaNat Immunol20089885786518604209
- ZhongZZhaiYLiangSTRPM2 links oxidative stress to NLRP3 inflammasome activationNat Commun20134161123511475
- MurakamiTOckingerJYuJCritical role for calcium mobilization in activation of the NLRP3 inflammasomeProc Natl Acad Sci U S A201210928112821128722733741
- VenegasCKumarSFranklinBSMicroglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s diseaseNature2017552768535536129293211
- ZhouRYazdiASMenuPTschoppJA role for mitochondria in NLRP3 inflammasome activationNature2011469732922122521124315
- LeeG-SSubramanianNKimAIThe calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMPNature2012492742712312723143333
- RossolMPiererMRaulienNExtracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptorsNat Commun20123132923271661
- SollbergerGStrittmatterGEKistowskaMFrenchLEBeerH-DCaspase-4 is required for activation of inflammasomesJ Immunol Baltim Md 19502012188419922000
- ElliottEISutterwalaFSInitiation and perpetuation of NLRP3 inflammasome activation and assemblyImmunol Rev20152651355225879282
- Muñoz-PlanilloRKuffaPMartínez-ColónGSmithBLRajendiranTMNúñezGK+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matterImmunity20133861142115323809161
- ShimadaKCrotherTRKarlinJOxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosisImmunity201236340141422342844
- IyerSSHeQJanczyJRMitochondrial cardiolipin is required for Nlrp3 inflammasome activationImmunity201339231132323954133
- NakahiraKHaspelJARathinamVAAutophagy proteins regulate innate immune response by inhibiting NALP3 inflammasome-mediated mitochondrial DNA releaseNat Immunol201112322223021151103
- GaidtMMEbertTSChauhanDHuman monocytes engage an alternative inflammasome pathwayImmunity201644483384627037191
- NakamuraYKambeNSaitoMMast cells mediate neutrophil recruitment and vascular leakage through the NLRP3 inflammasome in histamine-independent urticariaJ Exp Med200920651037104619364881
- MankanAKDauTJenneDHornungVThe NLRP3/ASC/Caspase-1 axis regulates IL-1β processing in neutrophilsEur J Immunol201242371071522213227
- NeteaMGNold-PetryCANoldMFDifferential requirement for the activation of the inflammasome for processing and release of IL-1β in monocytes and macrophagesBlood2009113102324233519104081
- PicciniACartaSTassiSLasigliéDFossatiGRubartelliAATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1β and IL-18 secretion in an autocrine wayProc Natl Acad Sci U S A2008105238067807218523012
- JamillouxYLefeuvreLMagnottiFFamilial Mediterranean fever mutations are hypermorphic mutations that specifically decrease the activation threshold of the Pyrin inflammasomeRheumatology201857110011129040788
- MisawaTTakahamaMKozakiTMicrotubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasomeNat Immunol201314545446023502856
- SubramanianNNatarajanKClatworthyMRWangZGermainRNThe adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activationCell2013153234836123582325
- YangC-SKimJ-JKimTSSmall heterodimer partner interacts with NLRP3 and negatively regulates activation of the NLRP3 inflammasomeNat Commun20156611525655831
- ElliottEIMillerANBanothBCutting edge: mitochondrial assembly of the NLRP3 inflammasome complex is initiated at primingJ Immunol Baltim Md 19502018200930473052
- LiXThomeSMaXMARK4 regulates NLRP3 positioning and inflammasome activation through a microtubule-dependent mechanismNat Commun201781598628656979
- WuY-HKuoW-CWuY-JParticipation of c-FLIP in NLRP3 and AIM2 inflammasome activationCell Death Differ201421345146124270411
- MariniESGiampietriCPetrungaroSThe endogenous caspase-8 inhibitor c-FLIPL regulates ER morphology and crosstalk with mitochondriaCell Death Differ20152271131114325501600
- ParkSJulianaCHongSThe mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activityJ Immunol201319184358436624048902
- GuanKWeiCZhengZMAVS promotes inflammasome activation by targeting ASC for K63-linked ubiquitination via the E3 ligase TRAF3J Immunol Baltim Md 195020151941048804890
- IchinoheTYamazakiTKoshibaTYanagiYMitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infectionProc Natl Acad Sci U S A201311044179631796824127597
- BryanNBDorfleutnerARojanasakulYStehlikCActivation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domainJ Immunol200918253173318219234215
- ZhangZMeszarosGHeW-TProtein kinase D at the Golgi controls NLRP3 inflammasome activationJ Exp Med201721492671269328716882
- Fernandes-AlnemriTWuJYuJ-WThe pyroptosome: a supra-molecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activationCell Death Differ20071491590160417599095
- HeYZengMYYangDMotroBNúñezGNek7 is an essential mediator of NLRP3 activation downstream of potassium effluxNature2016530759035435726814970
- Schmid-BurgkJLChauhanDSchmidtTA genome-wide CRISPR (clustered regularly interspaced short palindromic repeats) screen identifies NEK7 as an essential component of NLRP3 inflammasome activationJ Biol Chem2016291110310926553871
- ShiHWangYLiXNLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome componentNat Immunol201617325025826642356
- WangYYangCMaoKChenSMengGSunBCellular localization of NLRP3 inflammasomeProtein Cell20134642543123609011
- ShiC-SShenderovKHuangN-NActivation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destructionNat Immunol201213325526322286270
- SpalingerMRLangSGottierCPTPN22 regulates NLRP3-mediated IL1B secretion in an autophagy dependent mannerAutophagy Epub201788
- SongNLiuZ-SXueWNLRP3 phosphorylation is an essential priming event for inflammasome activationMol Cell201768118518728943315
- SaitoMFujisawaANishikomoriRSomatic mosaicism of CIAS1 in a patient with chronic infantile neurologic, cutaneous, articular syndromeArthritis Rheum200552113579358516255047
- HumphriesFBerginRJacksonRThe E3 ubiquitin ligase Pellino2 mediates priming of the NLRP3 inflammasomeNat Commun201891156029674674
- MayorAMartinonFDe SmedtTPétrilliVTschoppJA crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responsesNat Immunol20078549750317435760
- HanSLearTBJeromeJALipopolysaccharide primes the NALP3 inflammasome by inhibiting its ubiquitination and degradation mediated by the SCFFBXL2 E3 ligaseJ Biol Chem201529029181241813326037928
- HanSJeromeJAGregoryADMallampalliRKCigarette smoke destabilizes NLRP3 protein by promoting its ubiquitinationRespir Res2017181228056996
- GuglielmoASabraAElberyMA mechanistic insight into curcumin modulation of the IL-1β secretion and NLRP3 S-glutathionylation induced by needle-like cationic cellulose nanocrystals in myeloid cellsChem Biol Interact201727411228669709
- PyBFKimM-SVakifahmetoglu-NorbergHYuanJDeubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activityMol Cell201349233133823246432
- KawashimaAKarasawaTTagoKARIH2 ubiquitinates NLRP3 and negatively regulates NLRP3 inflammasome activation in macrophagesJ Immunol Baltim Md 195020171991036143622
- YanYJiangWLiuLDopamine controls systemic inflammation through inhibition of NLRP3 inflammasomeCell20151601–2627325594175
- GuoCXieSChiZBile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasomeImmunity201645480281627692610
- MortimerLMoreauFMacDonaldJAChadeeKNLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutationsNat Immunol201617101176118627548431
- StutzAKolbeC-CStahlRNLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domainJ Exp Med201721461725173628465465
- LuheshiNMGilesJALopez-CastejonGBroughDSphingosine regulates the NLRP3-inflammasome and IL-1β release from macrophagesEur J Immunol201242371672522105559
- SpalingerMRKasperSGottierCNLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22J Clin Invest201612651783180027043286
- SongHLiuBHuaiWThe E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3Nat Commun201671372727929086
- MaoKChenSChenMNitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shockCell Res201323220121223318584
- MishraBBRathinamVAKMartensGWNitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1βNat Immunol2013141526023160153
- RodgersMABowmanJWFujitaHThe linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activationJ Exp Med201421171333134724958845
- LeeJYSeoDYouJThe deubiquitinating enzyme, ubiquitin-specific peptidase 50, regulates inflammasome activation by targeting the ASC adaptor proteinFEBS Lett2017591347949028094437
- StehlikCKrajewskaMWelshKKrajewskiSGodzikAReedJCThe PAAD/PYRIN-only protein POP1/ASC2 is a modulator of ASC-mediated nuclear-factor-kappa B and pro-caspase-1 regulationBiochem J2003373Pt 110111312656673
- HaraHTsuchiyaKKawamuraIPhosphorylation of ASC acts as a molecular switch controlling the formation of speck-like aggregates and inflammasome activityNat Immunol201314121247125524185614
- LabbéKMcIntireCRDoironKLeblancPMSalehMCellular inhibitors of apoptosis proteins cIAP1 and cIAP2 are required for efficient caspase-1 activation by the inflammasomeImmunity201135689790722195745
- MeissnerFMolawiKZychlinskyASuperoxide dismutase 1 regulates caspase-1 and endotoxic shockNat Immunol20089886687218604212