Abstract
Background
Patients with cystic fibrosis (CF) experience recurrent infections and develop chronically infected lungs, which initiates an altered immunological alveolar environment. End-stage pulmonary dysfunction is a result of a long sequence of complex events in CF, progressing to alveolar macrophage dysfunction via a T-helper 2 (TH2) dominated alveolar inflammation with CD20 T-cell activation, induced by the chronic infection and showing a poor prognosis. There is great potential for treatment in transforming the TH2 into the more favorable T-helper 1 (TH1) response.
Methods
Current literature in the PubMed database and other sources was reviewed in order to evaluate aspects of the innate alveolar host defense mechanisms and the potential impact on the immunoinflammatory response of inhalation of granulocyte-macrophage colony-stimulating factor (GM-CSF) in patients with CF.
Results
It seems that the cellular host defense, (ie, the alveolar macrophage and neutrocyte function) and the inhaled GM-CSF interact in such a way that the so-called tolerant alveolar environment dominated by the TH2 response may be transformed into an active TH1 state with a normal pulmonary host defense. The shift of the TH2 to the TH1 subset dominated by specific and unspecific antibodies may be achieved after the inhalation of GM-CSF. A clinical report has shown promising results with inhalation of GM-CSF in a chronically-infected CF patient treated with several antibacterial and antifungal agents. Inhaled GM-CSF transformed the tolerance toward the Gram-negative infection reflected by the so-called TH2 subset into the more acute TH1 response characterized by recruitment of the T-cells CD8 and CD16, a condition related to better-preserved lung function. This indicated a transformation from a state of passive bacterial tolerance toward the Gram-negative infecting and colonizing bacteria. This GM-CSF effect cannot be achieved by administering the drug via the IV route because the drug is water-soluble and too large to penetrate the alveolocapillary membrane.
Conclusions
Inhalation of GM-CSF seems to be a novel way to positively modulate the alveolar environment toward an altered immunological state, reflected by a positive change in the pattern of surrogate markers, related to better preservation of pulmonary function and thus improved outcomes in CF patients. It is suggested that future studies examining standard endpoint variables such as number of infections and amount of antibiotics used should be supplemented by surrogate markers, to reveal any positive cellular and cytokine responses reflecting changes in the alveolar compartment after GM-CSF inhalation. The immunological alveolar environment should be monitored by a specific pattern of surrogate markers. Continued research is clearly indicated and the role of inhaled GM-CSF in modulating pulmonary host defense in CF patients should be investigated in a large study.
Introduction
Patients with cystic fibrosis (CF) are subject to recurrent and chronic lung infections due to impairment of the natural host defenses of their airways.Citation1 End-stage pulmonary dysfunction in CF is a result of a long sequence of recurrent infections and colonizations that induce alveolar macrophage dysfunction via a T-helper 2 cell (TH2) dominated alveolar inflammation.Citation2 The TH2 response is tolerant toward multiple infections.Citation3
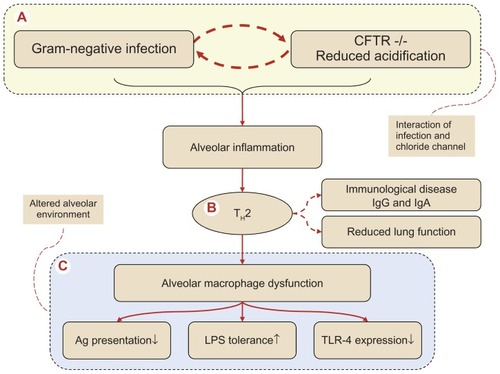
The cystic fibrosis transmembrane conductance regulator chloride channel (CFTR) plays a central role in bactericidal activity both in the neutrocyte and the macrophage. The defective CFTR−/− macrophages have reduced intracellular acidification and a Gram-negative infection initiates an alveolar TH2 response with B-cell activation resulting in antibody formation.Citation4 In this way the response is transformed from the normal alveolar host defense into the well characterized immunologic and allergic alveolar disorder accompanied by increased systemic immunoglobulin formation, which contributes to the pathogenesis of CF.Citation4 The TH2 response has been connected with an unfavorable and accelerated decline in lung function.Citation5 End-stage alveolar macrophage dysfunction is reflected by reduced antigen (Ag) presentation,Citation6 enhanced tolerance toward lipopolysaccharide (LPS),Citation1,Citation7 and reduced expression of recognition receptors, the so-called toll-like receptors (TLR), where TLR4 recognizes Gram-negatives.Citation8 The reduced alveolar host defense may also induce remodeling of the peripheral airways, leading to a fall in lung function which ultimately results in pulmonary failure.Citation9 This pathogenic step is only seen to affect the pulmonary host defense: circulating monocytes that are not yet exposed to interaction in the specific pulmonary milieu seem unaffected, even though all host cells are supposed to be affected equally by the congenital CFTR mutation.Citation10 Ongoing stimulation of the LPS binding receptor (CD14+) on the alveolar host cells causes receptor internalization and induces a state of LPS tolerance and subsequently hyperinflammation ().Citation1 The same unresponsiveness of the host cells is suspected in the sinuses, where a finding of chronic sinusitis with bacterial colonization is almost ubiquitous in the CF patient,Citation11 potentially serving as a bacterial reservoir leading to recolonization of the lungs after antibacterial eradication of the lung infection.Citation12
Figure 1 Pathogenesis of the complex series of events in cystic fibrosis (CF) progressing to alveolar macrophage dysfunction via a TH2 dominated alveolar inflammation. (A) Denotes the interaction between the defective cystic fibrosis transmembrane conductance regulator (CFTR)−/− macrophages in respect to intracellular acidification and Gram-negative infection, an interaction involving a T-cell switch (B) to an alveolar T-helper 2 response with induction of B-cell activation with antibody formation, transforming CF into an immunological allergic disease characterized by increased systemic immunoglobulin formation (IgG and IgE). The TH2 response has been connected with an unfavorable accelerated declined spirometry. The end stage of the altered alveolar environment (C) with alveolar macrophage dysfunction, reflected by (i) reduced antigen (Ag) presentation, (ii) enhanced tolerance toward lipopolysaccharide (LPS) and finally (iii) reduced expression of recognition receptors, the so-called toll-like receptors (TLR) where TLR-4, which recognizes Gram-negatives, is decreased. In the end, remodeling of peripheral airways will take place producing a reduced alveolar host defense.
The dominant microbiological etiology of the chronic infections is Pseudomonas aeruginosa, Staphylococcus aureus, Burkholderia cepacia complex, Achromobacter xylosoxidans and Mycobacterium abscessus/chelonae. These are frequently isolated from CF lungs.Citation13 Microorganisms such as P. aeruginosa find a niche in the alveolar environment due to a whole host of bacterial survival “strategies” – the so-called bacterial “stealth strategy” – including mucoid exopolysaccharide production and biofilm formation, evading both the host defense system and antibiotic therapy. Once the lungs are chronically infected it is impossible to eliminate the causative agent. The ongoing colonization induces antibiotic resistance,Citation14 which results in a continuous reduction of lung function due to destruction of the lung tissue induced by the excessive inflammation.Citation15 A high or rapidly increasing number of anti-Pseudomonas antibodies has been correlated to a poor prognosis, while CF patients with a low number of anti-Pseudomonas antibodies show an improved outcome with chronic P. aeruginosa lung infection.Citation16 TH-cells are subdivided into two subsets based on their cytokine pattern: TH1 pattern cells are characterized by IFN-γ, production and activation of macrophages, and induction of cellular T-cell responses. In contrast, the TH2 pattern produces IL-4, IL-5, IL-9, and IL-13 and is characterized by increased CD20 T-cells ().Citation17
Table 1 Comparison of two subsets TH1 and TH2 each consisting of T-cells and a corresponding cytokine pattern
Increased IL-4 and reduced IFN-γ release from peripheral blood mononuclear cells (PBMCs) stimulated with Pseudomonas antigen has been demonstrated.Citation18
Inhaled GM-CSF and the TH1 subset
The beneficial TH1 subset may be induced by GM-CSF: T-lymphocytes are recruited into the alveolus as are antigen-presenting alveolar macrophages. The TH2 subset causing the tolerance towards the ever-present Pseudomonas species in the alveolar environment is downregulated by the inhaled GM-CSF. This makes inhaled GM-CSF a highly interesting new drug for inhalation with respect to alveolar immunomodulation. In addition, inhaled GM-CSF stays in the alveolus with no spill-over to the circulation and thus has no systemic adverse effects.Citation19
Research has shown that chronically-infected CF patients with the highest IFN-γ and IL-4 production also have the best-preserved lung function, indicating a beneficial potential for the modulation of the TH1/TH2 balance.Citation10
Animal studies and modulation of the immune system
It has been documented that IFN-γ treatment of rats with chronic P. aeruginosa lung infection results in increased neutrophilic-induced pulmonary inflammation with less reactive mononuclear cells.Citation24
A key cell in initiating and controlling the T-helper cell response is the dendritic cell (DC). Unless the DCs present and interact, naive T-cells will not be activated.Citation25 The resting alveolar macrophage will only be transformed into a DC if GM-CSF is present to stimulate the surface receptor of the alveolar macrophages (the autocrine function).Citation26 It may be hypothesized that increased serum granulocyte colony-stimulating factor (G-CSF) could be the cause of the skewed TH1/TH2-ratio observed in CF. Moreover, several publications have reported that granulocyte GM-CSF can induce a TH1 dominated response through modulation of the DCs.Citation27,Citation28 Indeed, chronically-infected CF patients were observed to have a significantly decreased GM-CSF/G-CSF ratio as compared to CF patients without a chronic P. aeruginosa lung infection.Citation5 Furthermore, this GM-CSF ratio correlated to the IFN-γ release from peripheral blood mononuclear cells (PBMCs) and lung function, indicating a beneficial role for GM-CSF treatment in CF. GM-CSF is normally produced locally by the alveolar macrophages,Citation21 being the first line of defense when clearing bacteria.Citation29 The innate immune function of the alveolar macrophage is significantly influenced by subsequently acquired immune responses.Citation30 The concentration of GM-CSF in the alveolar space is of utmost importance for the pulmonary host defense system.
Treatment of CF patients has hitherto focused on intensive long-term, high-dose, broad-spectrum antibiotics administered intravenously and by inhalation, combined with interventions aiming at enhanced clearance of mucus, eg, with DNA-ase.
Inhalation versus intravenous administration
Recently a paper discussed whether recombinant proteins should be inhaled or administered systemically in order to reach an alveolar target.Citation31 In the context of the present review one may question which route of administration should be preferred, when one intervenes with GM-CSF.
It has been documented that inhaled GM-CSF increases the number and function of phagocytic cells obtained from bronchoalveolar lavage (BAL), but with only sparse and transient increase in the number of myeloid cells in circulation.Citation32 Conversely, when administered intravenously (IV) there was only a limited response in alveolar cellularity. It follows that the pulmonary innate host defense is separated from the systemic defense system with respect to GM-CSF, and that the agent does not penetrate from the systemic circulation to the alveolar space or vice versa.Citation32 IV administration of even relatively small molecules like recombinant antitrypsin does not reach the alveolar space, as shown by the fact that only 2% of the IV-administered drug reaches the lung space.Citation33 For larger molecules like the recombinant protein Activated Protein C (APC), it has been documented that administered intravenously, APC had no effect even in the inflamed lung.Citation34 However, when inhaled, APC achieved the expected effect in the alveolus, without any systemic adverse effects.Citation35,Citation36
GM-CSF neutralizing autoantibodies in pulmonary alveolar proteinosis (PAP) have led to a lack of phagocytic function of alveolar macrophages.Citation37 Subcutaneous administration of GM-CSF has been tried in PAP to improve macrophage function, but only inhaled GM-CSF had an effect.Citation38 The explanation is that anti-GM-CSF blocking autoantibodies are detected both in serum and BAL fluid from patients with PAP.Citation39
A major point is the question of whether the inhaled drug reaches the GM-CSF receptors of alveolar macrophages in the peripheral airways.
By using a so-called micropump nebulizer, sufficiently small respirable aerosol particles with a size of ~2.5 μm are produced, which means that a high degree of peripheral lung deposition is obtained.Citation40,Citation41 Inhaled GM-CSF also appears safe because aerosol therapy did not cause any adverse effects in either lung cancer or in patients with pulmonary alveolar proteinosis.Citation42–Citation44 Based on these findings GM-CSF treatment may therefore be best inhaled rather than administered systemically.
Immunomodulatory treatment
Treating CF patients with GM-CSF inhalation has shown promising results.Citation45 The drug was inhaled in a dose of 250 μg/day for 1 week followed by 14 days’ pause, for a total period of 3 months. The GM-CSF response was monitored by immunoglobulins IgG and IgE, cytokines, and spirometry (forced expiratory volume in 1 second, FEV1). Interestingly, it brought about a noteworthy cytokine increase, ie, an IFN-γ increase from PBMCs activated with bacterial antigen.
Inhalation of GM-CSF was well tolerated without any signs or symptoms of pulmonary adverse effects, such as bronchoconstriction. The treatment seemed safe. None of the described findings following inhalation were documented after systemic GM-CSF therapy: ie, no switch in T-helper cell pattern and no change in immunologic allergic variables or cytokine response. It was not possible to document any effect in the alveolar space when systemic administration of GM-CSF or other biologics was used. These findings support the concept that the inhaled GM-CSF transformed the TH2 dominated alveolar condition into a TH1-like response (), a suggestion which is also in keeping with the fact that a TH2 response is seen when Aspergillus antigens appear in CF patients.Citation22 During the period of inhaled GM-CSF intervention, a noteworthy increase in IFN-γ release of PBMCs after stimulation with Mycobacterium abscessus antigen was noted, a TH1 response which was not demonstrated during systemic GM-CSF administration.
Furthermore it has been documented that transgenic mice overexpressing GM-CSF in alveolar epithelial cells have an enhanced resistance towards influenza by activation of the alveolar immune mechanisms that depend on alveolar macrophages.Citation46 These mice were highly resistant to infection with laboratory and clinical influenza strains, including the pandemic swine H1N1 strain. The mice had reduced lung injury and reduced mortality after influenza infection. They also showed increased levels of TNF-α and evidence of mononuclear cell infiltrates after infection. The histological findings suggested that the mice mounted a rapid host-immune response to influenza infection. These results indicate that the host response reduced the viral burden and most importantly reduced the mortality of the infection, emphasizing the therapeutic potential of inhaled GM-CSF.
Should host defense or total lung function be used as endpoints?
It has been predicted that the TH1 subset, increased by inhaled GM-CSF, is related to a much more favorable FEV1 and vital capacity (VC) in the longer run.Citation5 Further, the change in FEV1 does not sufficiently and sensitively reflect changes in the peripheral airways, because small airways normally contribute less than 30% of the total airway resistance.Citation47 This implies that substantial abnormalities could arise in these airways before the FEV1 becomes abnormal.Citation48,Citation49 It thus appears that measurement of total lung function (FEV1 and VC) does not, over a longer time span, reflect the changes of the small airways and alveolar environmental changes with sufficient sensitivity during the chronic infectious state. This is a most important point in the documentation of changes induced by inhaled GM-CSF, because GM-CSF exerts its effect on the alveolar macrophages. It therefore seems irrelevant to measure total lung function variables in order to reflect modifications after interventions such as inhaled GM-CSF, because measurements of FEV1/VC in a much later, irreversible phase reflect restrictive pulmonary dysfunction, such as extensive irreversible structural remodeling of small airways, with widespread bronchiectases, cyst formation, mucoid impaction, atelectasis, fibrosis, and vascular changes.Citation50 Alternative methods reflecting the immunological changes that prevent irreversible lung function damage must therefore be applied. The measurement of total lung function should therefore be abandoned because it is irrelevant in respect to the prevention of fibrosis in the CF patient. Lung function testing should not be the monitor – on the contrary it is only the endpoint. Therefore the monitor should be the pathophysiologic variables that prevent lung fibrosis: the surrogate variables as shown in , which reflect the multiple efficacy aspects of inhaled GM-CSF.
Table 2 Overview of potential surrogate markers in relation to monitoring the efficacy of inhaled GM-CSF
Discussion
Treating chronic lung infections in patients with CF becomes an increasing challenge in spite of the increasing use of both systemic and inhaled administered antibiotics. CF patients develop intolerance and allergic reactions to a number of antibiotics, along with reduction of renal and hepatic function. Since lung tissue damage is highly associated with the inflammatory reaction induced by infection, adjunctive immuno-modulators have increasingly been considered as necessary treatment options in CF.Citation51
Since antibacterial and anti-inflammatory interventions have not led to the expected result, a paradigm shift is needed. Treatment with GM-CSF could be such a new paradigm because its mechanism of action aims at the upregulation of the alveolar host defense. Here, the functions of the alveolar macrophages are considered most important in respect to the alveolar host defense towards bacteria and fungi, because the alveolar environment is dominated by tolerance toward the Gram-negatives – the so-called T-helper subset TH2; a state which is characterized by the chronic invasion of CD20 cells into the alveolar space, which is reflected by the increased immunoglobulins IgG and IgE.Citation52 Inhalation of the growth factor GM-CSF causes a transformation into the TH1 dominated subset, a change which could upregulate the pulmonary host defense even in the chronically-infected CF patient.
Alveolar host defense and the alveolar environment
The Achilles heel of CF is the infected lung with reduced clearance of bacteria, fungi, and viruses, causing progressive lung disease associated with unremitting bacterial infection, predominantly P. aeruginosa.Citation53 The gene defect in CF affects macrophages globally due to the lack of CFTR regulation, reducing the important phagosomal acidification and causing a reduced bactericidal activity triggered by the altered alveolar microenvironment, as underlined by the fact that the CF patient does not have an increased rate of systemic infections.Citation54 Loss of intracellular acidification in CFTR-deficient alveolar macrophages could therefore be the explanation for the enhanced survival of bacterial loads within the phagosomal compartment.Citation55 This apparent discrepancy between reduced alveolar macrophage function and lack of global infections may seem puzzling. However, the interaction between CFTR-affected alveolar macrophages and Gram-negative pulmonary infection precipitates a regionally-reduced pulmonary host defense. Normally alveolar macrophages induce inflammatory cytokines after exposure to Gram-negative bacteria via the TLRs. However, in CF patients, TLR4 expression in the bronchial epithelium is significantly reduced compared to healthy control subjects, which is emphasized by the finding that the TLR4 surface expression is normalized after correction of the CFTR-negative cells.Citation8 The loss of CFTR function appears to decrease innate immune responses, possibly by altering the expression of TLR4 on airway epithelial cells, thereby contributing to chronic bacterial infection in CF airways.Citation8
The defective macrophage and the alveolar environment in the CF patient
In a mouse model of CF based on mice infected with LPS, a defect in CF macrophage function has been suggested. The innate GM-CSF expression was reduced in CF-like mice as compared to wild-type mice,Citation56 in line with the findings of increased mortality, and reduced phagocytosis and intracellular bacterial killing of Gram-negatives by alveolar macrophages in GM-CSF (−/−) mice. By administering GM-CSF, the documented changes were reversed.Citation57
Along with the documented dysfunctional macrophages of the CF lung, which could explain the reduced capacity to clear bacterial infecting agents, the end result could be an excessive chronic inflammatory response, the TH2 dominated response.
The alveolar cellular recruitment process may lead to either a TH1 or a TH2 cell response. The TH1 response enhances a cellular profile with cytotoxic T-cells (CD8+) and natural killer cells (CD16+ (NK-cells)), with a specific cytokine IFN-γ pattern,Citation10 whereas the TH2 response corresponds to a chronic hyperinflammatory state with a different cytokine profile and recruitment of CD20+ cells with production of immunoglobulins (), triggered by ongoing stimulation of the antigens mostly from Pseudomonas spp.Citation10
The chronic local inflammatory state is dominated by the TH2 response, with activation of CD20+ cells, increased production of antibodies, and development of tolerance towards Gram-negative infections.Citation4 With the alveolar tolerance towards Gram-negative bacteria, the CD14 cells characteristic of the TH2 subset induce irreversible lung fibrosis and pulmonary dysfunction typical of CF terminal respiratory insufficiency.Citation23
Effect of inhaled GM-CSF
Inhaled GM-CSF can potentially enhance the complex host defense of the alveolar environment, because the drug enhances macrophage function via multiple points of action. (i) The TLR4 s acting on Gram-negative bacteria are increasingly expressed after treatment with GM-CSF, which facilitates the recognition and clearing of Gram-negatives.Citation58 (ii) The drug increases alveolar antigen presentation by enhanced opsonization and intracellular killing of bacteria after GM-CSF administration.Citation19 (iii) The local GM-CSF has been shown to shift T-helper response toward the essential TH1 subset, because the recruited cytotoxic (CD8) and NK-cells (CD16) from the systemic circulation may clear the infection.Citation20,Citation21 (iv) Inactivation of pulmonary fibroblasts (CD14). These factors combine to produce a more favorable lung function outcome.Citation5
It also appears likely that bacterial clearance is improved by a T-helper cell switch from an immunological TH2 response to the favorable TH1 setting. It appears that such a shift takes place during and after the GM-CSF inhalation period as both IgG and IgE are reduced and specific IgE anti-Aspergillus antibodies are reduced during the inhalation period.Citation5
The antibody response against P. aeruginosa in CF is a marker of chronic infection, inflammation, and tissue damage. The very high sensitivity of the assays makes it possible to characterize patients with negative antibody assays as being free of chronic P. aeruginosa infection.Citation59 The TH2-like immunological response also encompasses an allergic component with increased immunoglobulins that is typical for allergic bronchopulmonary aspergillosis. This condition is common in CF, with an incidence of 10%. The condition is diagnosed by increased levels of IgE, Aspergillus-specific IgE and IgG, or precipitins.Citation60 Cohen-Cymberknoh et al found that treatment with prednisolone, which is supposed to be the treatment of choice, did not suppress the antibodies.Citation61 With GM-CSF inhalation, on the other hand, IgE fell to almost zero and the IgG leveled out. Further it appeared that macrophage function was enhanced, because A. fumigatus was not cultured and acid-fast bacteria could not be detected in the patients’ sputum for a period after the GM-CSF inhalation.
These findings strongly support the concept that inhaled GM-CSF transforms the TH2-dominated alveolar condition into a TH1-like response, which is in keeping with the fact that a TH2 response is seen when Aspergillus antigens appear in CF patients.Citation22 During the GM-CSF inhalation period, stabilization of lung function (FEV1) was noted.Citation45 However, changes in FEV1 do not sufficiently and sensitively reflect changes in the peripheral airways because small airways normally contribute less than 30% of the total airway resistance. Citation47 Therefore substantial abnormalities could arise in these airways before FEV1 values become abnormal.Citation48 It appears that measurements of FEV1 do not sensitively reflect changes in the environment of small airways in early stages of the disease – the point of attack of inhaled GM-CSF. Later however, FEV1 values reflect respiratory failure due predominantly to extensive irreversible structural remodeling of the small airways, with widespread bronchiectases, cyst formation, mucoid impaction, atelectasis, fibrosis, and vascular changes.Citation50 In future, measurements of FEV1 should be combined with immunological variables (ie, surrogate variables). Inhaled GM-CSF did not produce pulmonary adverse effects in any of the published papers documenting the adverse effects of GM-CSF, ie, neither wheezing nor signs of bronchoconstriction were registered.
Inhaled GM-CSF also appears safe because aerosol therapy did not cause any adverse effects in either lung cancer or in patients with pulmonary alveolar proteinosis.Citation42–Citation44
Surrogate markers reflecting the changes in the alveoli
shows the multiple points of action of inhaled GM-CSF as they relate to future studies, most of which are aimed at monitoring the TH2 to TH1 switch.
These variables could provide a new approach to studying alterations in the alveolar environment after an intervention such as treatment with inhaled GM-CSF.
A major issue in the antibacterial strategy is that biofilm formation protects bacteria from antibiotic action. In CF patients there is frequent colonization with P. aeruginosa and the mucoid isolate forms a biofilm of which a major component is alginate. This may quantitatively be measured in sputum (). The mucoid isolate is a Pseudomonas strain that constantly signals neighboring bacteria via pheromones to produce biofilm. These colony forming units (CFU) can produce biofilm by increasing the bacterial density by a process called quorum sensing (QS).Citation62
This adaptive “stealth” strategy counteracts the combined antibacterial action of antibiotics and the endogenous alveolar immune system.
Treatment of the mucoid strains has hitherto not been able to counteract the biofilm formation. Because inhaled GM-CSF can upregulate the peripheral airways host defense, it could, just by reducing the CFUs, reduce the amount of alginate in sputum. The in vitro QS test could prove to be a useful marker of the efficacy of GM-CSF inhalation.
Conclusion
Inhaled GM-CSF seems to be a promising new treatment to improve host defenses in patients with CF. The chronic alveolar immuno-inflammatory status with colonization is a typical TH2-dominated inflammatory response. Such a subset unfortunately appears to be tolerant of Gram-negative bacteria. After inhalation of GM-CSF, however, the T-helper cells can be transformed into the TH1 subset, as documented by reduced immunoglobulin IgG and IgE concentration in a recent study.Citation45 The IgE concentration, which is related to allergy, was also reduced after GM-CSF inhalation, indicated by an altered cytokine pattern that showed increased TNF, IL-1β, IFN-γ, and IL-10 concentration. Inhaled GM-CSF was regarded as the only effective administration route because IV-administered GM-CSF, like other biologics, does not pass the alveolocapillary membrane. This important fact makes it possible to achieve a direct effect on the alveolar immune inflammatory response without adverse effects in the systemic pool of immunoactive cells.
In order to elucidate the role of inhaled GM-CSF in a human controlled trial, at least in a short term study of 6 weeks, it is necessary to apply not only the classical primary endpoints but to supplement with changes in specific surrogate variables each reflecting a specific function of the host system. In such a study we find it less important to emphasize classical spirometry measurements as the primary endpoint. The expected alterations in lung function do not reflect the changes in peripheral airways and the alveolar environment after administration of inhaled GM-CSF. The overall purpose of such a study is to show that inhaled GM-CSF in chronically colonized CF patients modifies the immuno-inflammatory state and switches the TH2 subset into the favorable TH1 subset.
Continued research is clearly indicated and the role of inhaled GM-CSF in modulating the pulmonary host defense in CF patients should be investigated in a large controlled study.
Disclosure
Lars Heslet (LH) has shares in the pharmacompany Serendex ApS Copenhagen, Denmark, which holds a patent related to inhaled GM-CSF. LH has, however, not received reimbursements, fees, or funding from any organization relating to the content or the preparation of this manuscript. LH declares that he has no other competing interests. Steen Nepper-Christensen (SNC) and Christiane Bay (CB) declare that they have no competing financial interests related to the preparation or the content of the manuscript. All authors read and approved the final manuscript.
References
- AlexisNEMuhlebachMSPedenDBNoahTLAttenuation of host defense function of lung phagocytes in young cystic fibrosis patientsJ Cyst Fibros200651172516356787
- ChmielJFBergerMKonstanMWThe role of inflammation in the pathophysiology of CF lung diseaseClin Rev Allergy Immunol200223152712162106
- HartlDGrieseMKapplerMPulmonary T(H)2 response in Pseudomonas aeruginosa-infected patients with cystic fibrosisJ Allergy Clin Immunol2006117120421116387607
- GreenbergerPAImmunologic aspects of lung diseases and cystic fibrosisJAMA199727822192419309396654
- MoserCJensenPØPresslerTSerum concentrations of GM-CSF and G-CSF correlate with the Th1/Th2 cytokine response in cystic fibrosis patients with chronic Pseudomonas aeruginosa lung infectionAPMIS2005113640040915996157
- KnightRAKollnbergerSMaddenBYacoubMHodsonMEDefective antigen presentation by lavage cells from terminal patients with cystic fibrosisClin Exp Immunol199710735425479067530
- del FresnoCGómez-PinaVLoresVMonocytes from cystic fibrosis patients are locked in an LPS tolerance state: down-regulation of TREM-1 as putative underlying mechanismPLoS ONE200837e266718628981
- JohnGYildirimAORubinBKGruenertDCHenkeMOTLR-4-mediated innate immunity is reduced in cystic fibrosis airway cellsAm J Respir Cell Mol Biol201042442443119502387
- Peterson-CarmichaelSLHarrisWTGoelRAssociation of lower airway inflammation with physiologic findings in young children with cystic fibrosisPediatr Pulmonol200944550351119382221
- BrazovaJSedivaAPospisilovaDDifferential cytokine profile in children with cystic fibrosisClin Immunol2005115221021515885645
- CuylerJPMonaghanAJCystic fibrosis and sinusitisJ Otolaryngol19891841731752738999
- FiskerJvon BuchwaldCJohansenHKMicrobial biofilm in rhinosinusitis and cystic fibrosisUgeskr Laeg2011173641741921299934
- SaimanLSiegelJInfection control in cystic fibrosisClin Microbiol Rev2004171577114726455
- HansenCRPresslerTHøibyNEarly aggressive eradication therapy for intermittent Pseudomonas aeruginosa airway colonization in cystic fibrosis patients: 15 years’ experienceJ Cyst Fibros20087652353018693078
- NicholsDPKonstanMWChmielJFAnti-inflammatory therapies for cystic fibrosis-related lung diseaseClin Rev Allergy Immunol200835313515318546078
- CalumHMoserCJensenPØShiraiRHøibyNCytokine and surface receptor diversity of NK cells in resistant C3H/HeN and susceptible BALB/c mice with chronic Pseudomonas aeruginosa lung infectionAPMIS2003111989189714510646
- SpellbergBEdwardsJEJrType 1/Type 2 immunity in infectious diseasesClin Infect Dis20013217610211118387
- MoserCHougenHPSongZRygaardJKharazmiAHoibyNEarly immune response in susceptible and resistant mice strains with chronic Pseudomonas aeruginosa lung infection determines the type of T-helper cell responseAPMIS1999107121093110010660139
- EksiogluEAMahmoodSSChangMReddyVGM-CSF promotes differentiation of human dendritic cells and T lymphocytes toward a predominantly type 1 proinflammatory responseExp Hematol20073581163117117562355
- Gonzalez-JuarreroMHattleJMIzzoADisruption of granulocyte macrophage-colony stimulating factor production in the lungs severely affects the ability of mice to control Mycobacterium tuberculosis infectionJ Leukoc Biol200577691492215767289
- TrapnellBCWhitsettJAGm-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defenseAnnu Rev Physiol20026477580211826288
- KnutsenAPBelloneCKauffmanHImmunopathogenesis of allergic bronchopulmonary aspergillosis in cystic fibrosisJ Cyst Fibros200212768915463812
- MeneghinAHogaboamCMInfectious disease, the innate immune response, and fibrosisJ Clin Invest2007117353053817332880
- JohansenHKHougenHPRygaardJHøibyNInterferon-gamma (IFN-gamma) treatment decreases the inflammatory response in chronic Pseudomonas aeruginosa pneumonia in ratsClin Exp Immunol199610322122188565302
- UenoHKlechevskyEMoritaRDendritic cell subsets in health and diseaseImmunol Rev200721911814217850486
- GrunigGBanzAde Waal MalefytRMolecular regulation of Th2 immunity by dendritic cellsPharmacol Ther20051061759615781123
- CauxCDezutter-DambuyantCSchmittDBanchereauJGM-CSF and TNF-alpha cooperate in the generation of dendritic Langerhans cellsNature199236064012582611279441
- MariottiSSargentiniVMarcantonioCT-cell-mediated and antigen-dependent differentiation of human monocyte into different dendritic cell subsets: a feedback control of Th1/Th2 responsesFASEB J20082293370337918556459
- ZhangPSummerWRBagbyGJNelsonSInnate immunity and pulmonary host defenseImmunol Rev2000173395110719666
- MedzhitovRJanewayCJrInnate immunityN Engl J Med2000343533834410922424
- HesletLLook on the “air side” in pneumoniaCrit Care Med200937277477519325384
- RoseRMKobzikLDushayKThe effect of aerosolized recombinant human granulocyte macrophage colony-stimulating factor on lung leukocytes in nonhuman primatesAm Rev Respir Dis19921465 Pt 1127912861443885
- BrandPBeckmannHMaas EnriquezMPeripheral deposition of alpha1-protease inhibitor using commercial inhalation devicesEur Respir J200322226326712952258
- LiuKDLevittJZhuoHRandomized clinical trial of activated protein C for the treatment of acute lung injuryAm J Respir Crit Care Med2008178661862318565951
- HesletLAndersenJSSengeløvHDahlbäckBDalsgaard-NielsenJInhalation of activated protein C: A possible new adjunctive intervention in acute respiratory distress syndromeBiologics20071446547219707316
- WaerhaugKKuzkovVVKuklinVNInhaled aerosolised recombinant human activated protein C ameliorates endotoxin-induced lung injury in anaesthetised sheepCrit Care2009132R5119356243
- KitamuraTTanakaNWatanabeJIdiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factorJ Exp Med1999190687588010499925
- Rodríguez PortalJARodríguez BecerraESánchez GarridoAResponse to inhaled granulocyte-macrophage colony-stimulating factor in a patient with alveolar proteinosisArch Bronconeumol2009453150152 Spanish19286115
- IoachimescuOCKavuruMSPulmonary alveolar proteinosisChron Respir Dis20063314915916916009
- SangwanSAgostiJMBauerLAAerosolized protein delivery in asthma: gamma camera analysis of regional deposition and perfusionJ Aerosol Med200114218519511681650
- LuisettiMKronebergPSuzukiTPhysical properties, lung deposition modeling, and bioactivity of recombinant GM-CSF aerosolised with a highly efficient nebulizerPulm Pharmacol Ther201124112312720728558
- AndersonPMMarkovicSNSloanJAAerosol granulocyte macrophage-colony stimulating factor: a low toxicity, lung-specific biological therapy in patients with lung metastasesClin Cancer Res1999592316232310499599
- WylamMETenRPrakashUBSAerosol granulocyte-macrophage colony-stimulating factor for pulmonary alveolar proteinosisEur Respir J200627358559316507860
- RaoRDAndersonPMArndtCASWettsteinPJMarkovicSNAerosolized granulocyte macrophage colony-stimulating factor (GM-CSF) therapy in metastatic cancerAm J Clin Oncol200326549349814528078
- MoserCJensenPØPresslerTHøibyNAdjunctive treatment with GM-CSF of CF patients with severe Mycobacterium abscessus lung infectionThe 19th Annual North American Cystic Fibrosis ConferenceBaltimore MDOctober 20–23Pediatr Pulmonol2005Suppl 28: 239272
- HuangF-FBarnesPFFengYGM-CSF in the lung protects against lethal influenza infectionAm J Respir Crit Care Med2011184225926821474645
- CochraneGMBenatarSRDavisJCollinsJVClarkTJCorrelation between tests of small airway functionThorax19742921721784831522
- MacklemPTMeadJResistance of central and peripheral airways measured by a retrograde catheterJ Appl Physiol19672233954014960137
- HesletLLung function inhomogeneity. A study of the pattern of single breath nitrogen washout test and radio-spirometryDan Med Bull19833042422586347541
- RegameyNHilliardTNSaglaniSQuality, size, and composition of pediatric endobronchial biopsies in cystic fibrosisChest200713161710171717317731
- SharmaSJaffeADixonGImmunomodulatory effects of macrolide antibiotics in respiratory disease: therapeutic implications for asthma and cystic fibrosisPaediatr Drugs20079210711817407366
- SzeligaJDanielDSYangC-HGranulocyte-macrophage colony stimulating factor-mediated innate responses in tuberculosisTuberculosis (Edinb)200888172017928269
- HeijermanHInfection and inflammation in cystic fibrosis: a short reviewJ Cyst Fibros20054Suppl 23515970469
- ChapronJZuberBKanaanRManagement of acute and severe complications in adults with cystic fibrosisRev Mal Respir2011284503516 French21549905
- DiABrownMEDeriyLVCFTR regulates phagosome acidification in macrophages and alters bactericidal activityNat Cell Biol20068993394416921366
- BrusciaEMZhangP-XFerreiraEMacrophages directly contribute to the exaggerated inflammatory response in cystic fibrosis transmembrane conductance regulator−/− miceAm J Respir Cell Mol Biol200940329530418776130
- BallingerMNPaineR3rdSerezaniCHCRole of granulocyte macrophage colony-stimulating factor during gram-negative lung infection with Pseudomonas aeruginosaAm J Respir Cell Mol Biol200634676677416474098
- BozinovskiSJonesJEVlahosRHamiltonJAAndersonGPGranulocyte/macrophage-colony-stimulating factor (GM-CSF) regulates lung innate immunity to lipopolysaccharide through Akt/Erk activation of NFkappa B and AP-1 in vivoJ Biol Chem200227745428084281412208854
- PresslerTKarpatiFGranströmMDiagnostic significance of measurements of specific IgG antibodies to Pseudomonas aeruginosa by three different serological methodsJ Cyst Fibros200981374218835753
- BartonRCHobsonRPDentonMSerologic diagnosis of allergic bronchopulmonary aspergillosis in patients with cystic fibrosis through the detection of immunoglobulin G to Aspergillus fumigatusDiagn Microbiol Infect Dis200862328729118947811
- Cohen-CymberknohMBlauHShoseyovDIntravenous monthly pulse methylprednisolone treatment for ABPA in patients with cystic fibrosisJ Cyst Fibros20098425325719447081
- LeeBSchjerlingCKKirkbyNMucoid Pseudomonas aeruginosa isolates maintain the biofilm formation capacity and the gene expression profiles during the chronic lung infection of CF patientsAPMIS20111194–526327421492226