Abstract
Background:
Cerebral palsy is a nonprogressive motor impairment syndrome that has no effective cure. The etiology of most cases of cerebral palsy remains unknown; however, recent epidemiologic data have demonstrated an association between fetal neurologic injury and infection/inflammation. Maternal infection/inflammation may be associated with the induction of placental cytokines that could result in increased fetal proinflammatory cytokine exposure, and development of neonatal neurologic injury. Therefore, we sought to explore the mechanism by which maternal infection may produce a placental inflammatory response. We specifically examined rat placental cytokine production and activation of the Toll-like receptor 4 (TLR4) pathway in response to lipopolysaccharide exposure at preterm and near-term gestational ages.
Methods:
Preterm (e16) or near-term (e20) placental explants from pregnant rats were treated with 0, 1, or 10 μg/mL lipopolysaccharide. Explant integrity was assessed by lactate dehydrogenase assay. Interleukin-6 and tumor necrosis alpha levels were determined using enzyme-linked immunosorbent assay kits. TLR4 and phosphorylated nuclear factor kappa light chain enhancer of activated B cells (NFκB) protein expression levels were determined by Western blot analysis.
Results:
At both e16 and e20, lactate dehydrogenase levels were unchanged by treatment with lipopolysaccharide. After exposure to lipopolysaccharide, the release of interleukin-6 and tumor necrosis alpha from e16 placental explants increased by 4-fold and 8–9-fold, respectively (P < 0.05 versus vehicle). Conversely, interleukin-6 release from e20 explants was not significantly different compared with vehicle, and tumor necrosis alpha release was only 2-fold higher (P < 0.05 versus vehicle) following exposure to lipopolysaccharide. Phosphorylated NFκB protein expression was significantly increased in the nuclear fraction from placental explants exposed to lipopolysaccharide at both e16 and e20, although TLR4 protein expression was unaffected.
Conclusion:
Lipopolysaccharide induces higher interleukin-6 and tumor necrosis alpha expression at e16 versus e20, suggesting that preterm placentas may have a greater placental cytokine response to lipopolysaccharide infection. Furthermore, increased phosphorylated NFκB indicates that placental cytokine induction may occur by activation of the TLR4 pathway.
Introduction
Cerebral palsy is a group of nonprogressive motor impairment syndromes that manifest during fetal or early postnatal life. Although cerebral palsy was previously attributed to perinatal hypoxia, this accounts for only a small percentage of cerebral palsy infants. Epidemiological data have established that fetal or intra-amniotic infections are associated with induction of fetal proinflammatory cytokines and the development of neurologic injuryCitation1 in preterm and near-term infants.Citation2–Citation5 Despite these findings, the majority of cerebral palsy is still unexplained by either fetal infection or hypoxia.Citation6,Citation7
Whereas clinically significant fetal infection is uncommon, maternal infections (eg, pyelonephritis) occur both antepartum and intrapartum. Maternal infection/inflammation has been associated with fetal brain injury and offspring cerebral palsy. Maternal urinary tract infection increases the risk of cerebral palsyCitation8,Citation9 and this effect may be more significant when the exposure occurs at a preterm gestation.
Several mechanisms associate maternal infection with fetal inflammation. Direct transfer of bacteria may occur from the maternal bloodstream across the placenta into the fetal circulation. Early investigations showed that Gram-negative lipopolysaccharide does not cross human chorioamniotic membranes in vitro; however, in vivo studies have been contradictory. A low dose of iodinated lipopolysaccharide injected into mice is not detected in fetal tissues,Citation10 but at significantly higher doses, iodinated tracer is detected in both the placenta and fetal tissues.Citation11 Alternatively, direct transplacental transfer of maternal cytokines may occur across the human placenta. Although there does not appear to be any transfer of tumor necrosis alpha (TNF-α), interleukin (IL)-1 or IL-8, the ability of IL-6 to cross the placenta remains controversial.Citation12,Citation13 In the absence of transplacental passage of lipopolysaccharide or cytokines, it has been proposed that the placenta acts as a site of cytokine production, following lipopolysaccharide activation, or maternally derived cytokines.
Studies from our laboratory indicate that simulated maternal infection with lipopolysaccharide evokes fetal systemic, amniotic, and placental inflammatory responses, and most importantly, increased expression of proinflammatory cytokines in the fetal brain.Citation14 Toll-like receptors (TLR) are essential for an effective host cell response to lipopolysaccharide. Circulating lipopolysaccharide binding to membrane-bound TLR4 results in a series of kinase cascades that phosphorylate and translocate nuclear factor kappa light chain enhancer of activated B cells (NFκB) into the nucleus where it acts as a transcription factor stimulating production of proinflammatory cytokines by binding to promoter genes.Citation15–Citation17 Although an essential receptor for lipopolysaccharide signaling, TLR4 alone cannot confer lipopolysaccharide responsiveness. In addition to TLR4, TLR2 specificity also includes bacterial lipoproteins. Following bacterial infection, the current evidence suggests that TLR4 and TLR2 activate similar intracellular cascades. However, TLR4 may primarily promote a cytokine response, while TLR2 may result in cellular apoptosis.Citation18 Because both TLR4 and TLR2 are likely to activate the same intracellular cascade, we focused primarily on the downstream effects of lipopolysaccharide binding (NFκB) as opposed to the different TLRs. The TLR4 pathway has been described in the human placenta, but not in the rat placenta. Human and rat placentas share important structural similarities, because both are hemochorial with comparable permeability to a range of hydrophilic compounds. Our laboratory and others have demonstrated the ability of the rat placenta to produce a cytokine response to maternal lipopolysaccharide.Citation14,Citation19
To explore the mechanism of maternal inflammation-induced placental inflammatory response, we developed an ex vivo rat placental explant model, which includes both the junctional zone (site of hormone production) and the labyrinth zone (site of maternal-fetal exchange). This ex vivo explant model provides the opportunity to analyze the placental inflammatory response under multiple experimental conditions. The placental explant model maintains the structure of the placental tissues and keeps them in contact with their normal cellular environment, thereby facilitating physiological interactions. Although this model does not recreate in vivo homeostasis, it does allow greater control of the environment and provides for greater efficiency compared with animal models. Using this model, we examined TNF-α and IL-6 proinflammatory cytokine production in response to lipopolysaccharide. We specifically focused on these cytokines because previous in vivo studies from our laboratory demonstrated induction of TNF-α and IL-6 in maternal serum, amniotic fluid, chorioamnion, and placenta after maternal exposure to lipopolysaccharide but not IL-10 mRNA.Citation14 Additionally, we investigated the mechanism by which placental proinflammatory cytokine release occurs, by establishing the presence of TLR4 and quantifying nuclear phosphorylated NFκB with and without lipopolysaccharide. We hypothesized that maternal lipopolysaccharide activates the placental TLR4 signaling pathway, which results in induction and release of proinflammatory cytokines into the fetal compartment.
Materials and methods
Animals
Eight-week-old first-time pregnant Sprague-Dawley rats (230–240 g) in body weight, (Charles River Laboratories Inc, Hollister, CA) were housed in a facility with constant temperature and humidity, on a controlled 12-hour light/dark cycle and an ad libitum diet (controls) of standard laboratory chow (protein 23%, fat 4.5%, metabolizable energy 3030 kcal/kg; Lab Diet 5001, Gray Summit, MO). All studies were carried out in accordance with the established guidelines of the ethics committee of the Los Angeles Biomedical Research Institute at Harbor-UCLA Medical Center, the American Association for Accreditation of Laboratory Care, and National Institute of Health guidelines.
Explant cultures
At embryonic day 16 (e16, n = 6), and embryonic day 20 (e20, n = 7) the rats were sacrificed using an overdose of 4% isoflurane. Gestational sacs were removed from the uterus. Membranes attached to the placentas and deciduas were excised. The mid-portion of each placenta was divided into two explants (1 mm each) which were washed three times in phosphate-buffered saline. Two explants from each placenta were cultured in individual Costar Netwell supports (15 mm diameter, 74 μm mesh, Lowell, MA) in 2 mL of RPMI 1640 medium (Invitrogen, Carlsbad, CA) supplemented with 5% heat-inactivated fetal bovine serum, and 100 μg/mL Mycosin (Invivogen, Carlsbad, CA) and incubated in a standard Sanyo incubator (5% CO2 in air, 37°C) for 12 hours. After 12 hours, the culture media were replaced with media including different concentrations of lipopolysaccharide (0, 1, and 10 μg/mL, Sigma-Aldrich, St Louis, MO). After 30 minutes to 6 hours of incubation with lipopolysaccharide, the supernatants were collected, and stored at −80°C. The tissue explants were removed, weighed, and stored at −80°C. All explants and supernatants incubated with lipopolysaccharide 0 μg/mL are referred to as vehicles.
Explant integrity assessment
Lactate dehydrogenase is a cytoplasmic enzymeCitation20 which is rapidly released into cell culture medium upon membrane damage or cell lysis. Therefore, concentrations of lactate dehydrogenase in tissue explant and media assess placental membrane integrity during experiments. To assess membrane integrity in our explant cultures, we measured lactate dehydrogenase levels remaining in tissue explants and released into culture media after 6 hours of explant incubation (37°C) using a commercially available kit (Sigma-Aldrich). Briefly, tissue explants from e16 (n = 6) and e20 (n = 7) were homogenized in lactate dehydrogenase lysis solution using an ultrasonic cell disruptor (Ultrasonic Power Corporation, Freeport, IL), centrifuged, and the supernatants collected. Aliquots of homogenized tissue explants and culture supernatants were pipetted into separate wells of a 96-well plate with the lactate dehydrogenase mixture and incubated for 30 minutes. The reaction was terminated by addition of a 1/10 volume of 1 N HCl to each well. Lactate dehydrogenase activity was determined by measuring the increase in absorbance at 490 nm. Each assay was run with a known standard curve of lactate dehydrogenase (Sigma-Aldrich) in duplicate, and each treatment was performed in triplicate.
Enzyme-linked immunosorbent assay determinations
We determined supernatant levels of IL-6 and TNF-α proinflammatory cytokines because they are clinically relevant in maternal infection-mediated cerebral palsy.Citation21 We used commercially available enzyme-linked immunosorbent assay kits (R&D Systems, Minneapolis, MN) according to the manufacturer’s protocols. Briefly, for each cytokine, 50 μL of assay diluent and 50 μL of supernatant were pipetted into separate wells precoated with specific antibody for either rat IL-6 or TNF-α and incubated for 2 hours at room temperature, following which the wells were rinsed five times with a wash buffer (provided with the kits). Next, an enzyme-linked antibody specific for rat IL-6 or TNF-α was added to the wells for an additional 2 hours. After five washes with the wash buffer, a substrate solution was added to each well for 30 minutes, following which a colored product representing the level of cytokine in the supernatant was obtained. Levels of IL-6 and TNF-α in the supernatants were quantified by optical density readings at 450 nm. Each assay was run with known standards and each treatment was performed in triplicate.
Subcellular fractionation
Total and nuclear fractions were isolated using the method described by Hikim et al.Citation22 Next, e16 and e20 explants treated and not treated with lipopolysaccharide (n = 6 from each group) were sonicated on ice in buffer A [0.25 M sucrose, 50 mM HEPES, 10 mM NaCl, 10 mM ethylenediamine tetra-acetic acid, 2 mM DTT, 2 mM PMSF, one tablet protease inhibitor (complete mini tablets, Roche, Indianapolis, IL), and one tablet phosphatase inhibitor (PhosSTOP, Roche, Indianapolis, IL)], and incubated on ice for 15 minutes after which 100 μL of each lysate extract was transferred into a separate sterile Eppendorf tube (total protein fraction). The remaining crude homogenates were centrifuged at 1000 g for 10 minutes at 4°C to isolate the nuclear pellet which was redissolved in T-PER buffer (Thermo Scientific, Waltham, MA). The purity of the nuclear fractions was assessed by Western blotting using antibodies to mitochondrial cytochrome c oxidase subunit IV (COXVI, Cell Signaling Technology, Danvers, MA), and lysine-specific demethylase 1 (LSD1) (Cell Signaling Technology). Fractional protein concentrations were determined by bicinchoninic acid solution (Thermo Scientific). All fractions were frozen at −80°C.
TLR4 and phosphorylated NFκB Western blot analysis
Lipopolysaccharide treated or untreated total and nuclear protein lysates from e16 (55 μg) and e20 (55 μg) were separated on a 4%–12% gradient polyacrylamide gel (Invitrogen, Carlsbad, CA), and transferred to Immobilon-P membranes (BioRad, Hercules, CA). The membranes were incubated in a blocking solution (5% powdered nonfat milk in Tris buffered saline and 0.05% Tween-20 [TBS-T]) at room temperature for one hour, and then in the same solution containing either TLR4 (1:2500; Cell Signaling Technology) or phosphorylated NFκB (1:2500; Cell Signaling Technology) primary antibody overnight at 4°C. Following three 10-minute washes in TBS-T, the membranes were incubated with a secondary goat antirabbit polyclonal antibody for TLR4 (1:3000; Vector Laboratories, Burlingame, CA) or a rabbit antimouse monoclonal antibody for NF-κB (1:2000; Cell Signaling Technology) labeled with horseradish peroxidase. After washes with TBS-T, the blots were developed using a chemiluminescence Western blotting kit (Denville, Metuchen, NJ), and subsequently exposed onto X-OMAT Kodak film (Eastman-Kodak, Rochester, NY). For equal loading, the blots were stripped and reprobed with either β-actin antibody (1:2000, BioRad) for total protein or LSD1 antibody (1:1000; Cell Signaling Technology) for nuclear protein.
Statistical analysis
Differences between 0, 1, and 10 μg/mL lipopolysaccharide-treated groups were compared using one-way analysis of variance (IL-6, TNF-α, lactate dehydrogenase), and a two-tailed Student’s t-test (NFκB, TLR4) using SigmaStat (version 3.0; Systat Software Inc, Chicago, IL). Values were expressed as the mean ± standard error and considered statistically significant at P < 0.05.
Results
Explant integrity
Cell membrane integrity was assessed by comparing levels of lactate dehydrogenase in tissue and lactate dehydrogenase released into culture after 6 hours of treatment with and without lipopolysaccharide at e16 and e20. At e16, there was no significant difference in lactate dehydrogenase levels from tissue explants treated with 1 or 10 μg/mL lipopolysaccharide (2.2 ± 0.52 U and 1.5 ± 0.37 U lactate dehydrogenase/mL, respectively) compared with vehicle (2.4 ± 0.73 U lactate dehydrogenase/mL, P = 0.52). Similarly, in culture supernatants from explants treated with 1 or 10 μg/mL, levels of lactate dehydrogenase released into media (0.50 ± 0.06 and 0.36 ± 0.09 U lactate dehydrogenase/mL, respectively) were comparable with those from vehicle (0.48 ± 0.08 U lactate dehydrogenase/mL, P = 0.36). Likewise, at e20, there was no significant difference in lactate dehydrogenase levels from tissue explants treated with 1 or 10 μg/mL lipopolysaccharide (66.7 ± 15.9 U and 47.5 ± 6.4 U lactate dehydrogenase/mL, respectively) compared with vehicle (51.3 ± 11.7 U lactate dehydrogenase/mL, P = 0.21). In culture supernatants from explants treated with 1 or 10 μg/mL, the levels of lactate dehydrogenase released into culture media (5.6 ± 0.58 and 4.9 ± 0.55 U lactate dehydrogenase/mL respectively) were similar to those from the vehicle (3.9 ± 0.85 U lactate dehydrogenase/mL, P = 0.27).
Proinflammatory cytokine induction by lipopolysaccharide
At e16, explants exposed to lipopolysaccharide for 6 hours demonstrated a significant release in proinflammatory cytokines into media. Indeed, IL-6 release increased by 4.4-fold following exposure to 1 μg/mL (, 3845 ± 618 pg/mL/g tissue, P < 0.05), and by 4.9-fold after treatment with 10 μg/mL lipopolysaccharide (, 4276 ± 475 pg/mL/g tissue, P < 0.05) compared with vehicle (1167 ± 338 pg/mL/g tissue). TNFα release was increased by 8.2-fold after exposure to 1 μg/mL lipopolysaccharide (, 755 ± 110 pg/mL/g tissue, P < 0.05) and 9.4-fold after 10 μg/mL lipopolysaccharide treatment (, 869 ± 97 pg/mL/g tissue, P < 0.05) compared with the vehicle (92 ± 51 pg/mL/g tissue).
Figure 1 Fold change of pro-inflammatory cytokine interleukin (IL)-6 and tumor necrosis factor alpha (TNFα) released from preterm (e16) and near-term (e20) placental explants following lipopolysaccharide (LPS) treatments. Explants were incubated with increasing concentrations of LPS (0 μg/mL, 1 μg/mL and 10 μg/mL) for 6 hours. Enzyme-linked immunosorbent assay kits were used to determine supernatant levels of the cytokines IL-6 and TNFα. Explants exposed to 1 μg/mL and 10 μg/mL LPS (A) demonstrated a significant release in IL-6. *P < 0.05 versus vehicle. Preterm (e16) LPS treated explants had higher IL-6 induction than e20 treated explants. +P < 0.05 versus e20 at each matched dose (1 and 10 μg/mL). Also explants exposed to either 1 μg/mL or 10 μg/mL LPS (B) demonstrated a significant release in TNFα. *P < 0.05 versus vehicle. Preterm (e16) LPS treated explants had higher TNFα induction than e20 treated explants. +P < 0.05 versus e20 at each matched dose (1 and 10 μg/mL).
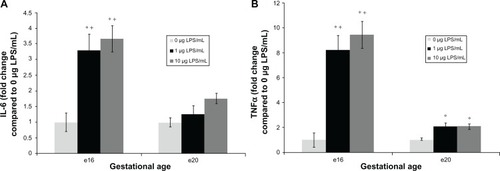
Conversely, at e20, proinflammatory cytokine IL-6 levels released from explants exposed to 1 or 10 μg/mL lipopolysaccharide were not significantly different from the vehicle. Thus, IL-6 levels increased by 1.25-fold after exposure to 1 μg/mL lipopolysaccharide (, 5303 ± 1118 pg/mL/g tissue), and by 1.75-fold after 10 μg/mL lipopolysaccharide (, 7371 ± 677 pg/mL/g tissue) compared with vehicle (4191 ± 568 pg/mL/g tissue, P=0.62). Notably, e16 lipopolysaccharide-treated explants had higher IL-6 induction than e20 treated explants at each matched dose (1 and 10 μg/mL, P < 0.05). Unlike e16, TNF-α release at e20 was increased by only twofold compared with the vehicle, although this difference was statistically significant for either 1 μg/mL (, 677 ± 87 pg/mL/g tissue, P < 0.05) or 10 μg/mL lipopolysaccharide exposure (, 674 ± 69 pg/mL/g tissue, P < 0.05) compared with the vehicle (324 ± 56 pg/mL/g tissue). Notably, e16 lipopolysaccharide-treated explants showed higher TNFα induction than e20 treated explants at each matched dose (1 and 10 μg/mL, P < 0.05).
TLR4 and NFκB protein expression
For TLR4 and NFκB proteins, we detected one immunoreactive band of the expected molecular weight on Western blots of placental protein lysates from specimens with and without lipopolysaccharide at e16 and e20 ( and ). Densitometric analysis showed that 1 μg/mL lipopolysaccharide did not change placental TLR4 expression in the total protein lysates by 6 hours at e16 () or by 2 hours at e20 ().
Figure 2 Toll-like receptor 4 expression from preterm (e16) and near-term (e20) placental explant protein lysates following LPS treatments. Explants were incubated with or without LPS (0 μg/mL and 1 μg/mL) for 6 hours. Western blot (A and B) and densitometric analysis (C) showed that TLR4 protein expression was unaffected by LPS exposure at both gestational ages. P = 0.85 (e16) and p = 0.87 (e20) vs vehicle (0 μg/mL).
Abbreviations: LPS, lipopolysaccharide; TLR, toll-like receptor.
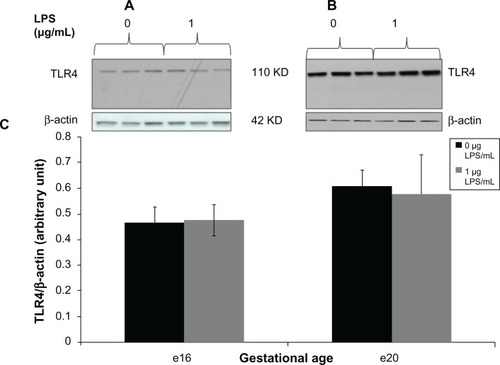
Figure 3 Phosphorylated nuclear NF-kB protein expression from preterm (e16) and near-term (e20) placental explants following LPS treatments. Explants were incubated with or without LPS (0 μg/mL and 1 μg/mL) for 30 minutes and 2 hours. Western blot (A and B) and densitometric analysis (C) showed that phosphorylated nuclear NF-κB was significantly increased in both e16 and e20 after exposure to LPS. +P = 0.064 vs untreated controls (0 μg/mL) (e16), and *P < 0.05 versus vehicle (0 μg/mL) (e16 and e20).
Abbreviations: NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; LPS, lipopolysaccharide.
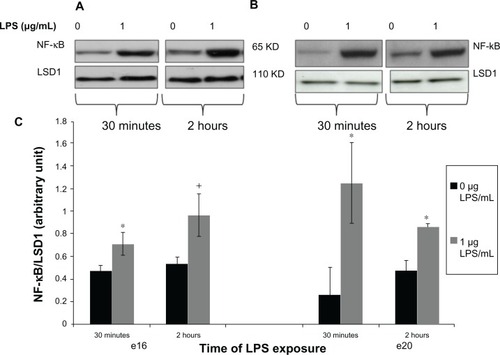
Upon activation, NFκB protein translocates from the cytoplasm to the nucleus where it activates proinflammatory genes. Our results demonstrated that nuclear phosphorylated NFκB increased in preterm placental explants after 30 minutes of lipopolysaccharide exposure (, P < 0.05 versus vehicle). After 2 hours of lipopolysaccharide exposure, nuclear phosphorylated NFκB expression was still greater in the lipopolysaccharide-treated explants versus untreated controls, but was no longer significant (). Likewise, for near-term, phosphorylated NFκB was increased in the nucleus placental explants after 30 minutes of lipopolysaccharide exposure (, P < 0.05 versus vehicle). After 2 hours of lipopolysaccharide exposure, nuclear phosphorylated NFκB expression remained greater in the lipopolysaccharide-treated explants (, P < 0.05 versus vehicle).
Discussion
In this study, exposure to lipopolysaccharide stimulated greater proinflammatory IL-6 and TNF-α cytokine release at e16 (preterm) compared with e20 (near-term) in rat placental explants. These findings suggest that the preterm placenta may be more susceptible to a lipopolysaccharide infection-induced placental cytokine response. The placental tissue exhibited constitutive TLR4 that was not influenced by lipopolysaccharide doses tested at either preterm or near-term gestation. After 30 minutes of lipopolysaccharide exposure, there was increased phosphorylated NFκB in the nucleus of the preterm and near-term explants, consistent with lipopolysaccharide-mediated activation of the TLR4 pathway.
Clinically, studies have linked maternal infection/inflammation with fetal brain injury and offspring cerebral palsy through fetal brain proinflammatory cytokines. The primary brain lesion associated with cerebral palsy is periventricular leukomalacia.Citation23 Proinflammatory cytokines TNF-α and IL-6 are expressed in 88% of neonatal brain specimens with periventricular leukomalacia, compared with only 18% of cases without periventricular leukomalacia.Citation24 Therefore, these cytokines may mediate the development of preterm neurologic injury because the less developed preterm brain may be more susceptible to damage.Citation25
There are also many other potential mediators of this inflammatory response. In addition to proinflammatory cytokines, lipopolysaccharide and other bacterial components can induce the release or production of antimicrobial peptides, chemokines, prostaglandins, leukotrienes, and complement,Citation26 which can result in the creation of reactive oxygen species.Citation27 One human study found that cord blood samples from infants with cerebral palsy showed increased concentrations of epidermal growth factor and three chemokines (B lymphocyte chemoattractant, MCP-3, and monokine induced by interferon gamma) compared with controls.Citation26 However, the majority of the clinical investigation into fetal neurologic injury and inflammatory response uses the release of proinflammatory cytokines as a marker for inflammation.Citation27 Therefore, we focused this study on these proinflammatory cytokines.
We found a greater induction of IL-6 and TNF-α in the preterm placenta versus the near-term placenta regardless of the concentration of lipopolysaccharide used. Previously, we analyzed the in vivo maternal inflammatory response 6 hours after lipopolysaccharide injection at e16 and e20,Citation19,Citation28,Citation29, and the maternal proinflammatory cytokine response to lipopolysaccharide was more than twofold greater at e16 versus e20. This emphasizes that both maternal compartments and the placenta induce more proinflammatory cytokines at a preterm gestational age than at near-term. In contrast with lipopolysaccharide-stimulated responses, basal cytokines in the untreated placenta were increased at e20 versus e16. Interestingly, in human pregnancy, IL-1β and IL-8 expression were increased in third trimester amnion and in the choriodecidual tissue,Citation30 and TNFα mRNA levels were significantly higher in the term placenta versus the preterm placenta in the absence of intrauterine infection.Citation31 Thus, in both rat and human pregnancy, higher basal cytokines in term gestational tissues may play a role in establishment of labor.Citation30
Consistent with our findings of an exaggerated cytokine response from preterm rat placentas, human preterm placentas also showed an exaggerated inflammatory response.Citation32 In particular, human placental cell cultures from noninfected preterm deliveries released significantly greater amounts of TNF-α and IL-1β than cultures from nonlaboring women at term.Citation32 Compared with the term placenta, amnion cells and placental villous tissues from the preterm placenta showed a more pronounced cytokine response.Citation33 Thus, induction of proinflammatory cytokines in the preterm placenta may result in increased fetal cytokine exposure and subsequent neurologic damage.
One proposed mechanism for the induction of proinflammatory cytokines in response to an infectious process is the binding of lipopolysaccharide to TLR4, that results in a series of kinase cascades that subsequently phosphorylate and translocate NFκB into the nucleus where it acts as a transcription factor. To date, placental TLRs have not been described in the rat placenta. In humans, TLR4 has been demonstrated in first trimester trophoblast cell lines,Citation34 and first trimester and term syncytiotrophoblasts.Citation15–Citation17 Importantly, TLR4 has been reported in Hofbauer cells (placental macrophages) in preterm human placenta from gestations with a history of chorioamnionitis.Citation35 Thus, maternal inflammatory mediators may evoke placental responses in trophoblast or macrophage cells via a TLR pathway. In this study, TLR4 was detected in both the preterm and near-term rat placenta, although the level of expression was not affected by lipopolysaccharide exposure at 6 hours. However, TLR4 pathway activation in the rat placenta resulted in increased nuclear phosphorylated NFκB. Notably, after 30 minutes of lipopolysaccharide exposure, there was increased phosphorylated NFκB in the nucleus of both preterm and near-term placental explants, consistent with lipopolysaccharide-mediated activation of the TLR4 pathway. This suggests that the TLR4 NFκB pathway is activated in response to lipopolysaccharide in the rat placenta and is possibly one potential mechanism by which the cytokine response is induced.
Contrary to expectations, 30 minutes of lipopolysaccharide exposure resulted in greater nuclear translocation of phosphorylated NFκB in the near-term compared with preterm placenta, although this did not result in a more robust TNF-α and IL-6 cytokine response at a later gestational age. Notably, after nuclear NFκB acts as a transcription factor and promotes production of TNF-α and IL-6 mRNA, there are a number of post-transcriptional mechanisms that provide rate-limiting controls on the amount of cytokine that is translated and released.Citation36,Citation37 TNF-α and IL-6 induction is regulated post-transcriptionally by mRNA binding proteins and microRNAs.Citation38 Adenylate-uridylate-rich elements in the 3′UTR of IL-6 and TNF-α mRNA dictate mRNA degradation.Citation33,Citation37 Perhaps e20 placenta has an enhanced ability to regulate the production of proinflammatory cytokines under infection conditions via post-transcriptional mechanisms. Additional experiments are required to dissect the transcriptional and post-transcriptional mechanisms through which NFκB may promote placental production of TNF-α and IL-6 cytokines, and clarify the divergent NFκB and cytokine responses in the preterm and near-term placenta.
There are many proinflammatory mediators and signaling pathways that may potentially lead to placental or fetal inflammation and eventually to fetal neurologic injury. A clear limitation of this study is the narrow focus on proinflammatory cytokines and their induction via the NFκB pathway. Further research is warranted to clarify the role of the other inflammatory mediators (eg, other interleukins, and nitrergic or prostanoid pathways) that likely contribute to placental and fetal inflammation and possibly to the development of fetal neurologic injury.
In our ex vivo rat placental explant model, viability of placental tissue was maintained for up to 6 hours, as assessed by lactate dehydrogenase measurement in tissue and supernatant from lipopolysaccharide-treated and lipopolysaccharide-nontreated explants at e16 and e20. Lactate dehydrogenase is a cytoplasmic enzyme, and minimal alteration in its concentration in the media further substantiates that placental membrane integrity was preserved during the experiments. In support of our findings, lactate dehydrogenase release from human placental explants treated with lipopolysaccharide (1 μg/mL) remained unchanged for 24 hours, indicating that the lipopolysaccharide concentration used was not toxic to the explants.Citation39 Consistent with these findings, any release of cytokines in our model was likely due to lipopolysaccharide induction rather than tissue damage. Higher placental basal lactate dehydrogenase release at e20 versus e16 was probably dependent on gestational age.
In summary, we have established an ex vivo model of placental cytokine production with lipopolysaccharide induction of NFκB. In vivo rat studies have shown that pretreatment with the anti-inflammatory cytokine, IL-10, significantly reduced the size of the brain lesions and blocks the exacerbating effect of IL-1β on brain lesions.Citation40,Citation41 Antioxidants, such as N-acetyl-cysteine, have also been shown to inhibit proinflammatory cytokine release in human fetal membranes,Citation42 and prevent upstream events leading to NFκB activation in cell culture.Citation43 Moreover, maternal N-acetyl-cysteine administration blocks placental and amniotic fluid cytokine responses to maternal lipopolysaccharide injection, and prevents the increase in fetal brain IL-6.Citation19 This explant model may be used to examine the role of the placental cytokine cascade in the efficacy of these anti-inflammatory therapeutic strategies.
Acknowledgements
This work was supported by a National Institute of Health grant (R01HD054751). We acknowledge Linda Day and Stacy Behare for their technical assistance.
Disclosure
The authors report no conflicts of interest in this work.
References
- WuYWColfordJMJrChorioamnionitis as a risk factor for cerebral palsy: a meta-analysisJAMA2000284111417142410989405
- WuYWSystematic review of chorioamnionitis and cerebral palsyMent Retard Dev Disabil Res Rev200281252911921383
- GretherJKNelsonKBMaternal infection and cerebral palsy in infants of normal birth weightJAMA199727832072119218666
- WuYWEscobarGJGretherJKCroenLAGreeneJDNewmanTBChorioamnionitis and cerebral palsy in term and near-term infantsJAMA2003290202677268414645309
- NeufeldMDFrigonCGrahamASMuellerBAMaternal infection and risk of cerebral palsy in term and preterm infantsJ Perinatol200525210811315538398
- HankinsGDSpeerMDefining the pathogenesis and pathophysiology of neonatal encephalopathy and cerebral palsyObstet Gynecol2003102362863612962954
- StrijbisEMOudmanIvan EssenPMacLennanAHCerebral palsy and the application of the international criteria for acute intrapartum hypoxiaObstet Gynecol200610761357136516738164
- SpinilloACapuzzoEStronatiMOmettoADe SantoloAAccianoSObstetric risk factors for periventricular leukomalacia among preterm infantsBr J Obstet Gynaecol199810588658719746379
- PolivkaBJNickelJTWilkinsJR3rdUrinary tract infection during pregnancy: a risk factor for cerebral palsy?J Obstet Gynecol Neonatal Nurs1997264405413
- AshdownHDumontYNgMPooleSBoksaPLuheshiGNThe role of cytokines in mediating effects of prenatal infection on the fetus: implications for schizophreniaMol Psychiatry2006111475516189509
- KohmuraYKirikaeTKirikaeFNakanoMSatoILipopolysaccharide (LPS)-induced intra-uterine fetal death (IUFD) in mice is principally due to maternal cause but not fetal sensitivity to LPSMicrobiol Immunol2000441189790411145270
- AaltonenRHeikkinenTHakalaKLaineKAlanenATransfer of proinflammatory cytokines across term placentaObstet Gynecol2005106480280716199639
- ZaretskyMVAlexanderJMByrdWBawdonRETransfer of inflammatory cytokines across the placentaObstet Gynecol2004103354655014990420
- GayleDABelooseskyRDesaiMAmidiFNunezSERossMGMaternal LPS induces cytokines in the amniotic fluid and corticotropin releasing hormone in the fetal rat brainAm J Physiol Regul Integr Comp Physiol20042866R1024R102914988088
- BeijarECMallardCPowellTLExpression and subcellular localization of TLR-4 in term and first trimester human placentaPlacenta2006272–332232616338476
- JohnstonMVHagbergHSex and the pathogenesis of cerebral palsyDev Med Child Neurol2007491747817209983
- HolmlundUCebersGDahlforsARExpression and regulation of the pattern recognition receptors toll-like receptor-2 and Toll-like receptor-4 in the human placentaImmunology2002107114515112225373
- AkiraSTakedaKToll-like receptor signallingNat Rev Immunol20044749951115229469
- BelooseskyRGayleDAAmidiFN-acetyl-cysteine suppresses amniotic fluid and placenta inflammatory cytokine responses to lipopolysaccharide in ratsAm J Obstet Gynecol2006194126827316389042
- SoorannaSROteng-NtimEMeahRRyderTABajoriaRCharacterization of human placental explants: morphological, biochemical and physiological studies using first and third trimester placentaHum Reprod199914253654110100006
- DammannOLevitonAMaternal intrauterine infection, cytokines, and brain damage in the preterm newbornPediatr Res1997421189212029
- HikimAPLueYYamamotoCMKey apoptotic pathways for heat-induced programmed germ cell death in the testisEndocrinology200314473167317512810573
- FungGBawdenKChowPYuVChorioamnionitis and outcome in extremely preterm infantsAnn Acad Med Singapore200332330531012854373
- YoonBHParkCWChaiworapongsaTIntrauterine infection and the development of cerebral palsyBJOG2003110Suppl 2012412712763129
- LevitonAPreterm birth and cerebral palsy: is tumor necrosis factor the missing link?Dev Med Child Neurol19933565535588504899
- KaukolaTSatyarajEPatelDDCerebral palsy is characterized by protein mediators in cord serumAnn Neurol200455218619414755722
- GotschFRomeroRKusanovicJPThe fetal inflammatory response syndromeClin Obstet Gynecol200750365268317762416
- AwadNKhatibNGinsbergYN-acetyl-cysteine (NAC) attenuates LPS-induced maternal and amniotic fluid oxidative stress and inflammatory responses in the preterm gestationAm J Obstet Gynecol2011204545021411055
- BelooseskyRGayleDARossMGMaternal N-acetylcysteine suppresses fetal inflammatory cytokine responses to maternal lipopolysaccharideAm J Obstet Gynecol200619541053105717000238
- ElliottCLLoudonJABrownNSlaterDMBennettPRSullivanMHIL-1beta and IL-8 in human fetal membranes: changes with gestational age, labor, and culture conditionsAm J Reprod Immunol200146426026711642674
- BenyoDFSmarasonARedmanCWSimsCConradKPExpression of inflammatory cytokines in placentas from women with preeclampsiaJ Clin Endocrinol Metab20018662505251211397847
- SteinbornAGunesHRoddigerSHalberstadtEElevated placental cytokine release, a process associated with preterm labor in the absence of intrauterine infectionObstet Gynecol1996884 Pt 15345398841213
- DumitruCDCeciJDTsatsanisCTNF-alpha induction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathwayCell200010371071108311163183
- AbrahamsVMBole-AldoPKimYMDivergent trophoblast responses to bacterial products mediated by TLRsJ Immunol200417374286429615383557
- KumazakiKNakayamaMYanagiharaISueharaNWadaYImmunohistochemical distribution of toll-like receptor 4 in term and preterm human placentas from normal and complicated pregnancy including chorioamnionitisHum Pathol2004351475414745724
- StamouPKontoyiannisDLPosttranscriptional regulation of TNF mRNA: a paradigm of signal-dependent mRNA utilization and its relevance to pathologyCurr Dir Autoimmun201011617920173387
- KheraTKDickADNicholsonLBMechanisms of TNF alpha regulation in uveitis: focus on RNA-binding proteinsProg Retin Eye Res201029661062120813201
- ZhaoWLiuMD’SilvaNJKirkwoodKLTristetraprolin regulates interleukin-6 expression through p38 MAPK-dependent affinity changes with mRNA 3′ untranslated regionJ Interferon Cytokine Res201131862963721457063
- LahamNBrenneckeSPRiceGEInterleukin-8 release from human gestational tissue explants: the effects of lipopolysaccharide and cytokinesBiol Reprod19975736166209282999
- MesplesBPlaisantFGressensPEffects of interleukin-10 on neonatal excitotoxic brain lesions in miceBrain Res Dev Brain Res20031411–22532
- DommerguesMAPatkaiJRenauldJCEvrardPGressensPProinflammatory cytokines and interleukin-9 exacerbate excitotoxic lesions of the newborn murine neopalliumAnn Neurol2000471546310632101
- LappasMPermezelMRiceGEN-acetyl-cysteine inhibits phospholipid metabolism, proinflammatory cytokine release, protease activity, and nuclear factor-kappaB deoxyribonucleic acid-binding activity in human fetal membranes in vitroJ Clin Endocrinol Metab20038841723172912679464
- Legrand-PoelsSManigliaSBoelaertJRPietteJActivation of the transcription factor NF-kappaB in lipopolysaccharide-stimulated U937 cellsBiochem Pharmacol19975333393469065737