Abstract
Introduction
The c-Jun N-terminal kinases (JNKs) are involved in signal transduction of inflammatory bowel diseases. The aim of this study was to examine the function of JNKs by using a low-dose dextran sulfate sodium (DSS) model in JNK1 knockout mice (Mapk8−/−), JNK2 knockout mice (Mapk9−/−), and wild-type controls (WT1, WT2).
Methods
The animals were evaluated daily using a disease activity index. After 30 days, the intestine was evaluated histologically with a crypt damage score. CD4+ and CD8+ cells were quantified using immunofluorescence. Analysis of tumor necrosis factor-α (TNFα), interleukin-6 (IL-6), and transforming growth factor β1 (TGFB1) expression was carried out using LightCycler® real-time polymerase chain reaction.
Results
Cyclic administration of low-dose DSS (1%) was not able to induce features of chronic colitis in Mapk8−/− WT2 mice. By contrast, DSS administration significantly increased the disease activity index in WT1 and Mapk9−/− mice. In Mapk9−/− mice, the crypt damage score and the number of CD4+ and CD8+ cells as features of chronic colitis/inflammation were also significantly elevated. Expression of TNFα, IL-6, and TGFB1 was not altered by the JNK knockout.
Conclusion
Administering DSS at a defined low concentration that is unable to induce colitis in WT animals leads to clinically and histologically detectable chronic colitis in Mapk9−/− mice. The reason for this disease-inducing effect resulting from the loss of JNK2 remains to be elucidated. Expression of TNFα, IL-6, and TGFB1 does not appear to be involved; proapoptotic JNK2 may prolong the activity of proinflammatory immune cells, leading to perpetuation of the inflammation.
Introduction
Inflammatory bowel diseases (IBDs) are disorders of unknown etiology, characterized by chronic, relapsing inflammation of the gastrointestinal tract. Inappropriate activation of the immune system leads to overproduction of immune cells, inflammatory cytokines, and tissue-destructive enzymes.Citation1–Citation3 Mediators of IBDs activate a variety of intracellular signaling cascades, such as mitogen-activated protein kinases (MAPKs). MAPKs are components of inflammatory and immunological signaling cascades that integrate various extracellular stimuli and initiate inflammatory cellular responses.Citation4,Citation5 The MAPK signal transduction pathway appears to be fundamental in the initiation of inflammatory responses.Citation6 Several subgroups have been identified within the MAPK family, including p42/44 extracellular signal-related kinase, Jun N-terminal kinase (JNK), and p38 MAPK.Citation7 The JNK class of enzymes comprises three main types: JNK1, JNK2, and JNK3. The first two are more ubiquitously distributed, whereas the third is confined to the central nervous system and cardiac myocytes.Citation1 JNK1 and JNK2 are involved in the signal transduction of IBDs, and they regulate the expression of proinflammatory cytokines.Citation6,Citation8,Citation9 For this reason, JNKs are considered to be targets for IBD therapy.
Previous studies of the function of JNK in vitro and in vivo have been limited by the lack of a specific inhibitor. In a pilot clinical trial in patients with Crohn’s disease, administration of CNI-1493, a synthetic guanylhydrazone that inhibits phosphorylation of both p38 MAPK and JNK, significantly decreased disease activity.Citation6 Hollenbach et alCitation10 and Assi et alCitation11 confirmed the beneficial effects of the p38 inhibitor SB203580 in both dextran sulfate sodium (DSS)-induced and 2,4,6-trinitrobenzenesulfonic acid–induced murine colitis, underscoring the importance of this pathway in gastrointestinal inflammation. SP600125 has been identified as a specific inhibitor of all three isoforms of JNK.Citation12,Citation13 The in vivo effects of SP600125 on inhibition of cytokine synthesis and protection against tissue injury have been evaluated in several animal models of inflammation.Citation12,Citation14,Citation15 A cell-penetrating antiproteinase-resistant peptide inhibitor of JNK (D-JNKI-1), derived from the JNK binding domain of JNK-interacting protein-1, was developed to competitively inhibit JNK binding to its substrates.Citation16 The peptide inhibitor D-JNKI-1 was more potent than the small-molecule inhibitor SP600125.Citation16 D-JNKI-1 reduced the number of apoptotic cells, indicating suppression of the mitochondrial mechanism of apoptosis.Citation17
The beneficial effects of JNK inhibitors have implicated JNK as a critical regulator in IBDs. However, the specificity of some of these inhibitors has been questioned.Citation18 Parallel studies using JNK gene knockout approaches have therefore provided valuable insights into the roles of JNK. In this context, Kontoyiannis et alCitation8,Citation9 underlined the important role of JNK in IBDs by genetic deletion of JNK2 in the tumor necrosis factor-α (TNFα)-mediated TNFαΔARE model of experimental colitis. JNK gene knockout led to alleviation of the TNFα-mediated inflammatory reaction.
Several animal models for research on intestinal inflammation have been developed. In 1990, Okayasu et alCitation19 described an experimental model of acute and chronic colitis in mice, induced by DSS. The pathological features resembled those of IBDs. DSS administered in drinking water produced weight loss and bloody diarrhea. Histopathologically, the changes were characterized by colonic mucosal inflammation and ulceration. The inflammation was worse in the left colon than in the right colon and cecum. Due to its ease of administration and the induction of a well-characterized and reproducible mucosal injury, DSS has been widely used in animal models for acute and chronic colitis.
In the present study, a low-dose DSS model of chronic experimental colitis was used. Mild chronic colitis was induced in JNK1 knockout mice (Mapk8−/−) and JNK2 knockout mice (Mapk9−/−). The study investigated the effect of JNK knockout on intestinal inflammation, T-cell immune responses, and expression of JNK-dependent cytokines.
Materials and methods
Animals
The experiments were conducted in female JNK1 knockout mice (Mapk8−/−), JNK2 knockout mice (Mapk9−/−), and wild-type controls (WT1 and WT2). All of the mice were 10–12 weeks of age at the start of the experiment (weight 20–25 g). The mice were housed three per cage in standard laboratory conditions and supplied with drinking water and food ad libitum (ssniff M-Z, 10 mm; ssniff Spezialdiäten, Soest, Germany). They were kept at the Institute of Pharmacology at Schleswig-Holstein University Hospital, Campus Kiel, Germany, for 14 days before the study was initiated. At this time, the mice were mature and the body weights among the different strains were comparable.
Transgenic mice deficient in JNK1 and JNK2 (Mapk8−/− and Mapk9−/−) were kindly provided by Roger J Davis (Program in Molecular Medicine, University of Massachusetts, Worcester, MA) and Richard A Flavell (Howard Hughes Medical Institute, Yale University School of Medicine, New Haven, CT), and bred at the Institute of Pharmacology at Schleswig-Holstein University Hospital. Genetic inactivation using targeted disruption of JNK1 and JNK2, resulting in Mapk8−/− and Mapk9−/− genotypes, has been described in detail elsewhere.Citation20–Citation22 In both genotypes, the deleted exons coded for the TPY motif of the activation loop and for the two alternatively spliced JNK1/2α and β subdomains.Citation20–Citation22
The Mapk8−/− and Mapk9−/− mice were back-crossed once with C57/Bl6 WT mice that had not been genetically altered, in order to obtain WT control animals for the investigations. These heterozygous F2 offspring were crossed to obtain F2 homozygous JNK knockout mice and homozygous WT controls. This F2 generation was used as parent founders to breed the JNK knockout and WT control mice for the animal experiments (F3 generation). This breeding approach minimizes the genetic strain variability of knockout/control animals, provides a greater number of animals, and meets the criteria of the international guidelines (Banbury Conference).Citation23
Genomic DNA was isolated from mouse tail tips using the Roche DNA High Pure PCR Template Preparation Kit (Roche Diagnostics, Manheim, Germany) in accordance with the manufacturer’s instructions. To genotype the experimental animals, specific polymerase chain reaction (PCR) primer sets of three primers were used for each genotype to distinguish the endogenous locus from targeted alleles, as previously described.Citation24 For genomic PCR, 4 μL genomic DNA, 2 μL 10 × PCR buffer, 1.6 μL 2.5 mM dNTPs, 0.2 μL Taq polymerase (Eppendorf, Hamburg, Germany; 5 U/μL), 2.7 μL aqua dest, and 2.5 μL of each primer (10 μM) were used (total volume 20 μL). Cycling conditions for PCR amplification were 95°C for 5 minutes and 32 cycles (for the JNK1 primer set) of 95°C for 60 seconds, 55°C for 60 seconds, and 72°C for 30 seconds, followed by a final extension at 72°C for 10 minutes. For PCR with the JNK2 primer set, the conditions were identical except for a reduction to 26 cycles. PCR products were blotted on a 1% agarose gel with electrophoresis.Citation25
Induction of mild chronic DSS colitis
The DSS concentration of 1% for induction of mild chronic colitis was determined in previous studies (data not shown) and is suitable for experimental protocols. The JNK1 knockout mice (Mapk8−/−) and JNK2 knockout mice (Mapk9−/−) and wild-type controls (WT1 and WT2) (n = 9 in each group) received three cycles of DSS treatment (molecular weight 36–44 kDa; MP Biomedicals, Aurora, OH, USA), each consisting of 1% (w/v) DSS for 5 days in distilled water. Each cycle of DSS treatment was followed by a 5-day interval with distilled water only. After completion of the third DSS cycle (after 30 days), the animals were killed by decapitation in deep anesthesia. The control groups for each strain (n = 9 in each group) received distilled water without DSS, and were also sacrificed after 30 days.
Assessment of disease activity
Disease activity was quantified in all of the animals daily using the disease activity index (DAI) described elsewhere.Citation26–Citation29 The mice were weighed daily and visually inspected for stool consistency, diarrhea, and rectal bleeding. Hemoccult tests were performed daily to detect occult blood in the stool.
Tissue samples
After the mice had been sacrificed, the entire colon was dissected out. It was rinsed with normal saline and divided into four anatomical parts: cecum, proximal colon, mid-colon, and distal colon. All of the dissection steps were carried out in ice, and the tissue was snap-frozen in liquid nitrogen and stored at −80°C.
Histological evaluation of inflammation
For light microscopy, 7-μm transverse cryostat sections (rotary cryostat microtome; Leica Microsystems, Nussloch, Germany) from the four anatomical parts (cecum, proximal colon, mid-colon, and distal colon) of each animal were stained with hematoxylin and eosin. The slides were examined under a Leica DM4000 B microscope and were evaluated independently by two investigators (EH and AMM). The degree of inflammation was graded semiquantitatively, with blinding to the respective group. The severity of mucosal injury was defined using the crypt damage score (CDS), as described previously.Citation24,Citation26,Citation27,Citation30 With regard to intra-observer agreement, differences were noted in only 5% of the observations. Any differences in grading were resolved by a joint examination that included a third blinded examiner (AMC).
Evaluation of colonic cytokine expression using real-time PCR
mRNA expression of TNFα, transforming growth factor β1 (TGFB1), and interleukin-6 (IL-6) was evaluated as previously described.Citation26 Total RNA was prepared from specimens of the colon of JNK2 knockout mice (Mapk9−/−) and WT controls using the RNeasy Mini Kit and Shredder Kit (Qiagen, Hilden, Germany). DNA digestion was carried out using the RNase-Free DNase Set (Quiagen). Thereafter, RNA was submitted to a second DNAse digestion and transcribed using Promega M-MLV Reverse Transcriptase (Promega, Mannheim, Germany). Real-time PCR was performed using the LightCycler® System with Fast-Start DNA Master SYBR Green I (Roche Diagnostics). β-Actin was used as a reference gene. Calibrator-normalized relative quantification without efficiency correction was carried out using the LightCycler® Relative Quantification software, following the manufacturer’s instructions. After normalization, a multiplication factor was used to adjust the final calibrator-normalized relative ratios to reasonable values.
The following primers were used:
β-actin, 5′-ACGTTGACATCCGTAAAG-3′ (upstream) and 5′-CAGTAACAGTCCGCCT-3′ (downstream) (amplicon 286 bp);
TNFα, 5′-CATCTTCTCAAAATTCGAGTGACAA-3′ (upstream) and 5′-TGGGAGTAGACAAGGTACAA CCC-3′ (downstream) (amplicon 175 bp);
TGFB1, 5′-TGACGTCACTGGAGTTGTACGG-3′ (upstream) and 5′-GGTTCATGTCATGGATGGTGC-3′ (downstream) (amplicon 170 bp);
IL-6, 5′-GAGGATACCACTCCCAACAGACC-3′ (upstream) and 5′-AAGTGCATCATCGTTGTT CATACA-3′ (downstream) (amplicon 141 bp).
Immunofluorescence of activated lymphocytes
Immunofluorescence was carried out in 6-μm transverse cryostat sections from the distal colon (rotary cryostat microtome; Leica Microsystems). The sections were mounted on poly-L-lysine slides, air-dried, and fixed in methanol at room temperature. They were then blocked with Aurion BSA-c (Aurion Immuno Gold Reagents and Accessories, Wageningen, The Netherlands). Sections were subsequently incubated with the primary antibodies for 2 hours at room temperature: monoclonal rat anti-CD4 antibody (BD Biosciences, Heidelberg, Germany) diluted 1:400 (0.2 mg/mL) in phosphate-buffered saline (PBS) and monoclonal rat anti-CD8 antibody (BD Biosciences) diluted 1:100 (0.2 mg/mL) in PBS. After washing in PBS, tissue-bound antibodies were detected using Cy3-conjugated donkey anti-rat IgG (Dianova, Hamburg, Germany) diluted 1:100 (1.5 mg/mL). Incubation with the secondary antibody was carried out for 2 hours at room temperature and then at 4°C overnight. Sections were then washed in PBS. Each section had its own control using the secondary antibody only. Finally, the sections were mounted and analyzed using a fluorescence microscope (Leica DM4000 B microscope). Quantification of CD4+ and CD8+ cells was performed by examining two randomly selected high-power fields (with a magnification of 40,000×).
Statistical evaluation
The results for the DAI and CDS were expressed as means ± standard error of the mean (SEM). DAI values were analyzed using repeated-measures ANOVA over all time points. Comparison of CDS values and cytokine expression between the groups was carried out using one-way ANOVA; pair comparisons were conducted using Tukey’s test. The labeling density of CD4+ and CD8+ cells was evaluated with the t-test. P-values of ≤ 0.05 were considered statistically significant. Survival rates and hazard ratios were analyzed using a log rank test and Cox regression analysis, with P-values of ≤ 0.05 considered statistically significant; 95% confidence intervals were also indicated.
Statement of animal care
All of the animal experiments were performed in accordance with German legislation for the protection of animals and the US National Institutes of Health (NIH) guidelines for the use and care of laboratory animals. The experiments were all approved by the Ministry of Agriculture and the Environment (Ministerium fuer Landwirtschaft und Naturschutz), Kiel, Germany (V742-72241.121-22(10-2(05))).
Results
Genotyping of JNK knockout and wild-type controls
Genotyping of JNK-deficient mice and WT controls using PCR and gel electrophoresis confirmed the respective genotype in all of the experimental animals (data not shown).
Development of mild colitis in Mapk9−/− as opposed to Mapk8−/− mice
One of the nine animals (11.1%) died in the Mapk8−/− group and three of the nine WT1 animals (33.3%) died after treatment with DSS. None of the animals in the H2O control groups died. The differences in the mortality rates were not significant.
Differences in DAI values in the animals that survived up to the end point of the study (30 days) were statistically evaluated using repeated-measures ANOVA over all time points. As illustrated in , only WT1 animals developed significant chronic colitis, as assessed by the DAI, following cyclic administration of low-dose DSS (1.0%). The DAI was significantly higher in the WT1 DSS group, in comparison with the WT1 H2O control group (4.2 ± 0.7 vs 1.2 ± 0.3; P < 0.001). By contrast, in Mapk8−/− mice, cyclic administration of low-dose DSS (1.0%) was unable to induce chronic colitis with a significantly elevated DAI, in comparison with the H2O control group (3.2 ± 0.2 vs 1.6 ± 0.5; P = 0.056) (). There were no significant differences in DAI values between the WT1 and Mapk8−/− mice following DSS treatment.
Figure 1 The disease activity index (DAI) after administration of three cycles of 1.0% dextran sulfate sodium (DSS) or H2O as the control (means ± SEM).
Notes: (A) After administration of 1.0% DSS, the DAI in WT1 mice was significantly elevated, in comparison with the H2O control group (P ≤ 0.05). Cyclic administration of DSS in Mapk8−/− animals resulted in mild triphasic colitis, with no significant differences in comparison with the H2O control group or the WT1 DSS group (statistical evaluation: ANOVA over all time points). (B) After administration of 1.0% DSS in the WT2 animals, the DAI was not significantly elevated, in comparison with the H2O control group. Cyclic administration of DSS in Mapk9−/− animals resulted in mild triphasic colitis, which was significantly greater, in comparison with the H2O control group (P ≤ 0.001) and in comparison with the WT2 DSS group (P ≤ 0.05) (statistical evaluation: ANOVA over all time points).
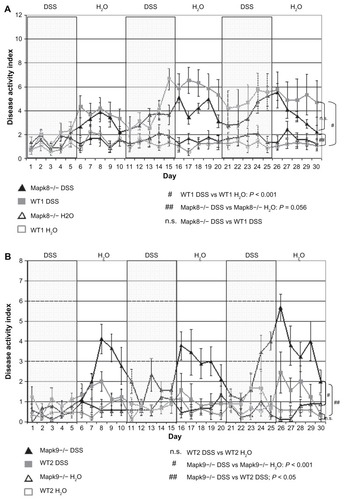
None of the animals in the Mapk9−/− and WT2 groups died (mortality 0%). Administration of 1.0% DSS in the WT2 animals did not induce chronic colitis, as reflected in a mean DAI of 1.1 (±0.4), which was not significantly elevated, in comparison with the value of 0.6 (±0.2) in the H2O control group. By contrast, cyclic administration of DSS in the Mapk9−/− animals resulted in mild triphasic colitis with a mean DAI of 2.3 (±0.3), which was significantly higher, in comparison with the H2O control group at 0.6 (±0.2) (P ≤ 0.001), and in comparison with the WT2 DSS group (P ≤ 0.05) ().
Histological scoring of the degree of inflammation
After DSS treatment, histological scoring using the CDS developed by Egger et alCitation24,Citation30 showed gradually elevated crypt damage in the medial and distal colon in Mapk8−/− mice (CDS in the mid-colon 2.6 ± 1.9 and in the distal colon 4.0 ± 2.4) and WT1 mice (CDS in the mid-colon 1.8 ± 1.2 and in the distal colon 5.1 ± 4.0). However, these differences in CDS did not reach statistical significance ().
Figure 2 The crypt damage score (CDS) after administration of three cycles of 1.0% dextran sulfate sodium (DSS) or H2O as the control (means ± SEM).
Notes: The most affected regions were the mid-colon and distal colon. (A) Differences in the CDS in JNK1 knockout (Mapk8−/−) and wild-type controls (WT1) did not reach statistical significance (CDS value comparison: one-way ANOVA; pair comparisons: Tukey’s test). (B) In the DSS-treated Mapk9−/− group, the CDS was significantly elevated, in comparison with the DSS-treated WT2 group (mid-colon, P = 0.034; distal colon, P = 0.047) and with the H2O control group (mid-colon, P = 0.001; distal colon, P = 0.047) (CDS value comparison: one-way ANOVA; pair comparisons: Tukey’s test).
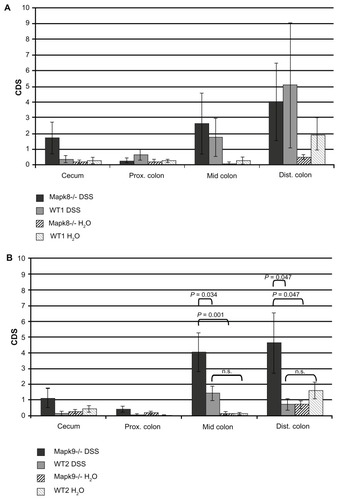
In Mapk9−/− mice, cyclic administration of 1% DSS also led to significant colitis in the mid-colon (CDS 4.0 ± 1.2) and distal colon (CDS 4.6 ± 1.9). In the DSS-treated Mapk9−/− group, the CDS was significantly elevated, in comparison with the DSS-treated WT2 group (mid-colon, P = 0.034; distal colon, P = 0.047) and in comparison with the H2O control groups (mid-colon, P = 0.001; distal colon, P = 0.047). DSS treatment of WT2 mice did not result in significant CDS changes, in comparison with H2O treatment ( and ).
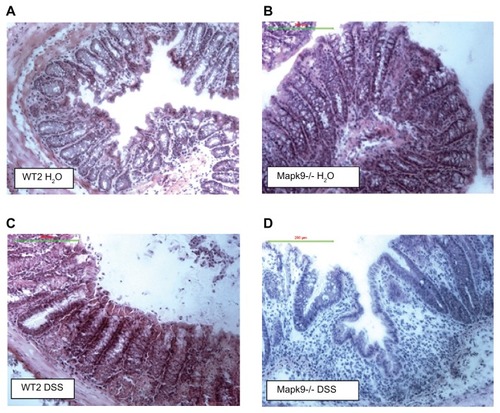
Figure 3 Hematoxylin–eosin staining of the distal colon.
Notes: (A) Wild-type controls (WT2) treated with H2O: normal mucosa with a regular crypt architecture. (B) JNK2 knockout mice (Mapk9−/−) treated with H2O: normal mucosa with a regular crypt architecture. (C) Wild-type controls (WT2) treated with three cycles of 1.0% dextran sulfate sodium (DSS): mild changes in the crypt architecture. (D) JNK2 knockout mice (Mapk9−/−) treated with three cycles of 1.0% DSS: severe changes in the crypt architecture, with ulceration and loss of crypts.
Inducing mild DSS colitis in JNK knockout mice did not alter the expression profile of cytokines TNFα, TGFB1, and IL-6
The expression profile of the cytokines TNFα, TGFB1, and IL-6 was examined in all of the animal groups (). Calibrator-normalized quantitative real-time PCR did not show any significant differences, either between the four Mapk8−/− WT1 groups or between the four Mapk9−/− WT2 groups in the experiment, with regard to the expression of TNFα, TGFB1, or IL-6.
Table 1 Relative mRNA expression (mean ± SEM) for TNFα, TGFB1, and IL-6
Activated lymphocytes in the inflamed colon
In the Mapk8−/− WT1 group, differences in the CDS did not reach statistical significance (). Analysis of the expression of CD8+ and CD4+ cells also did not show any notable differences.
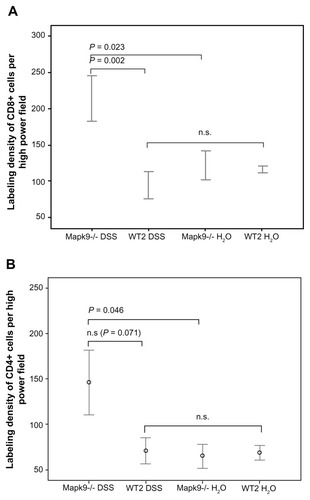
In the Mapk9−/− WT2 group, immunofluorescence analysis of CD4 and CD8 expression was carried out in tissue from the distal colon, as this part of the colon developed the highest grade of inflammation. After induction of chronic DSS colitis, CD8 staining was significantly higher in Mapk9−/− animals, in comparison with WT2 mice (P = 0.002). CD8 staining was also significantly increased in Mapk9−/− animals treated with DSS, in comparison with Mapk9−/− animals treated with H2O ( and ). Although the number of CD4+ cells was lower than the total number of CD8+ cells, immunofluorescence analysis of CD4 also showed significantly greater staining in Mapk9−/− animals treated with DSS, in comparison with Mapk9−/− animals treated with H2O. In DSS-treated Mapk9−/− animals, CD4 staining was greater, but did not show any significant differences, in comparison with DSS-treated WT2 mice (P = 0.071) ().
Figure 4 Immunofluorescence (Cy3-conjugated antibody) of CD8+ cells in the distal colon.
Notes: (A) Wild-type controls (WT2) treated with H2O. (B) JNK2 knockout mice (Mapk9−/−) treated with H2O. (C) Wild-type controls (WT2) treated with three cycles of 1.0% dextran sulfate sodium (DSS). (D) JNK2 knockout mice (Mapk9−/−) treated with three cycles of 1.0% DSS.
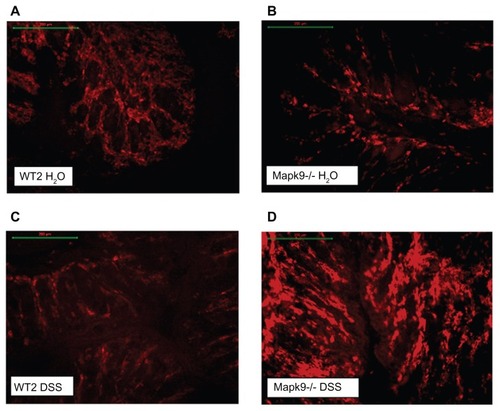
Figure 5 The labeling density of CD8 and CD4 (cells per high-power field) after administration of three cycles of 1.0% dextran sulfate sodium (DSS) or H2O as the control.
Notes: (A) After induction of chronic DSS colitis, CD8 staining was significantly higher in Mapk9−/− animals, in comparison with WT2 mice (P = 0.002). CD8 staining was also significantly elevated in Mapk9−/− animals treated with DSS, in comparison with Mapk9−/− animals treated with H2O (P = 0.023) (statistical evaluation: t-test). (B) There was significantly greater staining in Mapk9−/− animals treated with DSS, in comparison with Mapk9−/− animals treated with H2O (P = 0.046). In DSS-treated Mapk9−/− animals, CD4 staining was greater, but did not show any significant differences in comparison with DSS-treated WT2 mice (P = 0.071) (statistical evaluation: t-test).
Discussion
JNKs have a central position in the intracellular signaling that orchestrates inflammatory responses. Finding a way of inhibiting JNKs is therefore an attractive option in the treatment of inflammatory diseases, including IBDs. The present study investigated the function of the JNK isoforms JNK1 and JNK2 after induction of mild chronic colitis in JNK1 knockout mice (Mapk8−/−) and JNK2 knockout mice (Mapk9−/−). The clinical and histological features of inflammation were scored using the DAI and CDS, as described previously.Citation24,Citation30 As JNKs have multiple roles in T-cell immune responses and cytokine production,Citation21,Citation31 T-cell infiltration was evaluated using immunofluorescence, and the mRNA expression of the inflammatory cytokines TNFα, TGFB1, and IL-6 was quantified.
The observation that WT2 animals did not develop colitis after cyclic administration of low-dose DSS (1%), whereas WT1 animals developed chronic colitis, reflects a genetically determined differential susceptibility to DSS-induced colitis.Citation32 The concentration of 1% DSS was apparently too low to induce chronic colitis in WT2 animals. A further increase in the DSS dosage to 1.7%, as described by our group in a previous study, resulted in chronic colitis even in WT2 animals.Citation27 Cyclic application of 1.7% DSS triggered severe colitis in Mapk8−/− mice, Mapk9−/− mice, and wild-type controls, with DAI values between 4 and 11 and CDS values between 5 and 13. Unfortunately, the development of severe chronic colitis following cyclic application of 1.7% DSS was associated with high mortality rates of 75% in the Mapk9−/− group and 33% in the WT2 group. This limited the interpretation of histological differences between Mapk9−/− and WT2, due to the reduced number of surviving mice. For the same reasons, the further investigation of cytokines, CD4, and CD8 expression carried out in the present study could not be performed.Citation27
The intention in the present study was therefore to reduce the DSS dosage and achieve mild colitis with lower mortality rates. However, cyclic administration of 1% DSS in WT2 animals did not induce chronic colitis, reflected by a mean DAI of 1.1, which was not significantly elevated, in comparison with the H2O control group. In the Mapk9−/− animals, the same DSS dosage resulted in mild triphasic colitis with a mean DAI of 2.3, which was significantly higher, in comparison with the H2O control group at 0.6 (P ≤ 0.001) and in comparison with the WT2 DSS group (P ≤ 0.05) (). The reason for this disease-inducing effect from the loss of JNK2 remains to be elucidated.
The fact that one of the nine animals in the Mapk8−/− group died and three of the nine animals in the WT1 died after treatment with DSS, whereas none of the animals in the Mapk9−/− WT2 groups died, may also reflect a genetically determined differential susceptibility to DSS-induced colitis in Mapk8−/− WT1 animals, in comparison with Mapk9−/− WT2 animals.Citation32 Although the variability between mice was relatively high and the number of animals was limited (n = 9), inactivation of JNK2 resulted in a significant elevation of DAI and CDS, whereas there was no significant difference in Mapk8−/− mice, in comparison with WT1 mice after three cycles of 1% DSS administration. Although the DAI and CDS values were higher in WT1 mice, no significant differences were observed, either in histologic inflammation or in cytokine expression. One possible reason for this might be the experimental protocol used, in which a very mild form of chronic colitis was induced.
The distribution pattern of CD4+ and CD8+ cells reflected the degree of inflammation. The incidence of CD8+ cells was significantly higher in highly inflamed colon sections from Mapk9−/− mice, in comparison with WT2 mice and Mapk9−/− animals treated with H2O. These results are in accordance with the immune responses of JNK1-deficient and JNK2-deficient mice following infection with lymphocytic choriomeningitis virus.Citation33 In contrast to Mapk8−/−, which reduced the virus load, lymphocytic choriomeningitis virus infection generated more virus-specific CD8+ cells in Mapk9−/−, in comparison with the respective wild type. These results indicate that the JNK1 and JNK2 signal pathways have distinct roles in T-cell responses. JNK1 is involved in the survival of activated T cells during immune responses, and JNK2 plays a role in the control of CD8+ cell expansion. Previous studies showed that the absence of JNK2 causes increased proliferation of CD8+ cells, whereas JNK1-deficient CD8+ cells were unable to undergo antigen-stimulated expansion.Citation34 Experimental research on CD4+ cells revealed that the JNK signaling pathway is also required for differentiation of CD4+ cells into effector cells.Citation21,Citation35 The present data on CD4 expression do not support this conclusion, as increased CD4 labeling was found in the inflamed colon of Mapk9−/− mice. CD4+ cell expression was lower than CD8+ cell expression, but the difference did not reach significance.
The mechanisms that abnormally and persistently activate inflammation, leading to ongoing tissue damage, are varied and complex. Cytokines also play a central regulatory role.Citation2,Citation3 JNK has been implicated as an important regulator of the coordinated release of cytokines by immunocompetent cells and of the response of neutrophils to inflammatory stimuli. It positively regulates a variety of genes involved in inflammation, such as the TNFα, IL-1, and IL-6 genes.Citation36,Citation37 Inhibition of JNK activation effectively modulates TNFα secretion from macrophages and mast cells.Citation38,Citation39 However, JNK-dependent expression of the inflammatory cytokines TNFα, IL-6, and TGFB1 apparently did not play a major role in the present model of mild chronic DSS colitis. Calibrator-normalized quantitative real-time PCR did not show any significant differences, either between the four JNK1 groups or between the four JNK2 groups in the experiment, with regard to the expression of TNFα, TGFB1, or IL-6. This finding is in line with observations by Alex et al,Citation40 who reported an effect of these cytokines in acute DSS colitis, but not in chronic DSS colitis. An important aspect of further investigations by our study group will be the expression of other cytokines, such as interferon-γ (IFN-γ), IL-13, and IL-17, in order to obtain more information about the involvement of the Th1 and Th2 pathways in the exacerbated DSS colitis observed in JNK knockout mice.
The JNK branch of the MAPK signaling cascade has been implicated in the regulation of apoptosis in a variety of mammalian cell types.Citation22,Citation25,Citation41,Citation42 Apoptosis was not assessed in the present study. One possible hypothesis is that the proinflammatory effects caused by genetic deletion of JNK2 may be a consequence of inhibited JNK2-induced apoptosis. It might be speculated that functional deletion of proapoptotic JNK2 may prolong the activity of proinflammatory immune cells, leading to perpetuation of the inflammation. In addition, the increased inflammation in Mapk9−/− mice may be induced by blockage of the anti-inflammatory potency of the JNK2 isoform in these animals.Citation33,Citation34 No differences were observed in Mapk8−/− mice with regard to the degree of mucosal inflammation, in comparison with WT1 mice. A possible explanation for this might be that mice that are deficient in JNK1 are not able to develop the antiapoptotic activity of activated JNK1 that is achieved by transforming growth factor-β-associated kinase 1.Citation43 In view of the observation that the DSS colitis observed in JNK2 mice was not driven by the cytokines TNFα, TGFB1, or IL-6 that were being investigated, further studies are necessary in order to investigate the role of apoptosis in this process of chronic inflammation.
Deletion of JNK2 resulted in a deterioration of colitis in Mapk9−/− mice after DSS administration at a defined low concentration (1.0%). These results are in contrast to the expectations raised in studies that have reported a decrease in inflammation following administration of JNK inhibitors. It has been reported that the MAPK inhibitor CNI-1493 is able to alleviate human Crohn’s disease, even in some cases in which anti-TNFα antibody has failed.Citation6 It has also been demonstrated that p38 MAPK inhibition using SB203580 was effective in reducing disease in both DSS-induced and trinitrobenzenesulfonic acid-induced murine colitis. Both the clinical and the histological scores were significantly improved by treatment with SB203580.Citation10 Assi et alCitation11 reported that the JNK inhibitor SP600125 reduced infiltration of inflammatory cells, macrophage infiltration, and epithelial cell apoptosis. The observation in the present study that JNK2 deficiency leads to more severe colitis, whereas the JNK inhibitors described in the current literature have an anti-inflammatory effect, may be explained by the diverse functions of JNK signaling pathways in cellular metabolism and immunological regulation. It is very likely that these functions of JNK are needed for immunocompetence in both normal and pathogenic conditions. Genetic deletion of JNK2 is not equivalent to inhibition eg, by drugs. The former may result in compensatory (embryonic) changes whereas the pharmaceutical inhibition interferes with the acute situation only.
Conclusion
In contrast to the pharmacological inhibition of JNK, the present study shows that genetic deletion of JNK2 aggravates DSS colitis. It may be speculated that apoptosis of T cells is reduced, which is reflected in the high expression of CD4+ and CD8+ cells in the inflamed colon in DSS-treated Mapk9−/− mice. This might result in perpetuation of chronic intestinal inflammation. Further research is needed to confirm the relevance of apoptosis in this context.
Disclosure
The authors report no conflicts of interest in this work.
References
- ManningAMDavisRJTargeting JNK for therapeutic benefit: from junk to gold?Nat Rev Drug Discov20032755456512815381
- NeurathMFPetterssonSMeyer zum BuschenfeldeKHStroberWLocal administration of antisense phosphorothioate oligonucleotides to the p65 subunit of NF-kappa B abrogates established experimental colitis in miceNat Med19962999810048782457
- WaetzigGHSeegertDRosenstielPNikolausSSchreiberSp38 mitogen-activated protein kinase is activated and linked to TNF-alpha signaling in inflammatory bowel diseaseJ Immunol2002168105342535111994493
- RobinsonMJCobbMHMitogen-activated protein kinase pathwaysCurr Opin Cell Biol1997921801869069255
- SegerRKrebsEGThe MAPK signaling cascadeFaseb J1995997267357601337
- HommesDvan den BlinkBPlasseTInhibition of stress-activated MAP kinases induces clinical improvement in moderate to severe Crohn’s diseaseGastroenterology2002122171411781274
- MitsuyamaKSuzukiATomiyasuNPro-inflammatory signaling by Jun-N-terminal kinase in inflammatory bowel diseaseInt J Mol Med200617344945516465391
- KontoyiannisDBoulougourisGManoloukosMGenetic dissection of the cellular pathways and signaling mechanisms in modeled tumor necrosis factor-induced Crohn’s-like inflammatory bowel diseaseJ Exp Med2002196121563157412486099
- KontoyiannisDPasparakisMPizarroTTCominelliFKolliasGImpaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologiesImmunity199910338739810204494
- HollenbachENeumannMViethMRoessnerAMalfertheinerPNaumannMInhibition of p38 MAP kinase- and RICK/NF-kappaB-signaling suppresses inflammatory bowel diseaseFaseb J200418131550155215289440
- AssiKPillaiRGomez-MunozAOwenDSalhBThe specific JNK inhibitor SP600125 targets tumour necrosis factor-alpha production and epithelial cell apoptosis in acute murine colitisImmunology2006118111212116630028
- BennettBLSasakiDTMurrayBWSP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinaseProc Natl Acad Sci U S A20019824136811368611717429
- EshraghiAAWangJAdilEBlocking c-Jun-N-terminal kinase signaling can prevent hearing loss induced by both electrode insertion trauma and neomycin ototoxicityHear Res20072261–216817717098385
- EynottPRNathPLeungSYAdcockIMBennettBLChungKFAllergen-induced inflammation and airway epithelial and smooth muscle cell proliferation: role of Jun N-terminal kinaseBr J Pharmacol200314081373138014623764
- HanZBoyleDLChangLc-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritisJ Clin Invest20011081738111435459
- ZhuangZYWenYRZhangDRA peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenanceJ Neurosci200626133551356016571763
- MilanoGMorelSBonnyCA peptide inhibitor of c-Jun NH2-terminal kinase reduces myocardial ischemia-reperfusion injury and infarct size in vivoAm J Physiol Heart Circ Physiol20072924H1828H183517158645
- BogoyevitchMATherapeutic promise of JNK ATP-noncompetitive inhibitorsTrends Mol Med200511523223915882611
- OkayasuIHatakeyamaSYamadaMOhkusaTInagakiYNakayaRA novel method in the induction of reliable experimental acute and chronic ulcerative colitis in miceGastroenterology19909836947021688816
- DongCYangDDWyskMWhitmarshAJDavisRJFlavellRADefective T cell differentiation in the absence of Jnk1Science19982825396209220959851932
- YangDDConzeDWhitmarshAJDifferentiation of CD4+ T cells to Th1 cells requires MAP kinase JNK2Immunity1998945755859806643
- BrechtSKirchhofRChromikASpecific pathophysiological functions of JNK isoforms in the brainEur J Neurosci200521236337715673436
- WolferDPCrusioWELippHPKnockout mice: simple solutions to the problems of genetic background and flanking genesTrends Neurosci200225733634012079755
- EggerBCareyHVProcaccinoFReduced susceptibility of mice overexpressing transforming growth factor alpha to dextran sodium sulphate induced colitisGut199843164709771407
- KuanCYYangDDSamanta RoyDRDavisRJRakicPFlavellRAThe Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain developmentNeuron199922466767610230788
- ChromikAMMullerAMAlbrechtMOral administration of taurolidine ameliorates chronic DSS colitis in miceJ Invest Surg200720527328217972215
- ChromikAMMullerAMKornerJGenetic deletion of JNK1 and JNK2 aggravates the DSS-induced colitis in miceJ Invest Surg2007201233317365404
- CooperHSMurthySNShahRSSedergranDJClinicopathologic study of dextran sulfate sodium experimental murine colitisLab Invest19936922382498350599
- MurthySNCooperHSShimHShahRSIbrahimSASedergranDJTreatment of dextran sulfate sodium-induced murine colitis by intracolonic cyclosporinDig Dis Sci1993389172217348359087
- EggerBProcaccinoFLakshmananJMice lacking transforming growth factor alpha have an increased susceptibility to dextran sulfate-induced colitisGastroenterology199711338258329287974
- RinconMFlavellRADavisRJSignal transduction by MAP kinases in T lymphocytesOncogene200120192490249711402343
- MahlerMBristolIJLeiterEHDifferential susceptibility of inbred mouse strains to dextran sulfate sodium-induced colitisAm J Physiol19982743 Pt 1G544G5519530156
- ArbourNNanicheDHomannDDavisRJFlavellRAOldstoneMBc-Jun NH(2)-terminal kinase (JNK)1 and JNK2 signaling pathways have divergent roles in CD8(+) T cell-mediated antiviral immunityJ Exp Med2002195780181011927625
- ConzeDKrahlTKennedyNc-Jun NH(2)-terminal kinase (JNK)1 and JNK2 have distinct roles in CD8(+) T cell activationJ Exp Med2002195781182311927626
- DongCDavisRJFlavellRASignaling by the JNK group of MAP kinases. c-jun N-terminal KinaseJ Clin Immunol7200121425325711506194
- DerijardBHibiMWuIHJNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domainCell1994766102510378137421
- KallunkiTSuBTsigelnyIJNK2 contains a specificity-determining region responsible for efficient c-Jun binding and phosphorylationGenes Dev1994824299630078001819
- IshizukaTTeradaNGerwinsPMast cell tumor necrosis factor alpha production is regulated by MEK kinasesProc Natl Acad Sci U S A19979412635863639177222
- SwantekJLCobbMHGeppertTDJun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) is required for lipopolysaccharide stimulation of tumor necrosis factor alpha (TNF-alpha) translation: glucocorticoids inhibit TNF-alpha translation by blocking JNK/SAPKMol Cell Biol19971711627462829343388
- AlexPZachosNCNguyenTDistinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitisInflamm Bowel Dis200915334135218942757
- DietrichNThastrupJHolmbergCJNK2 mediates TNF-induced cell death in mouse embryonic fibroblasts via regulation of both caspase and cathepsin protease pathwaysCell Death Differ200411330131314685158
- KaiserRALiangQBuenoOGenetic inhibition or activation of JNK1/2 protects the myocardium from ischemia-reperfusion-induced cell death in vivoJ Biol Chem200528038326023260816043490
- SannaMGda Silva CorreiaJDucreyOIAP suppression of apoptosis involves distinct mechanisms: the TAK1/JNK1 signaling cascade and caspase inhibitionMol Cell Biol20022261754176611865055