
Abstract
Purpose
Following major trauma, genes involved in adaptive immunity are downregulated, which accompanies the upregulation of genes involved in systemic inflammatory responses. This study investigated microRNA (miRNA)-mRNA interactome dysregulation in circulating T cells of patients with major trauma.
Patients and Methods
This study included adult trauma patients who had an injury severity score ≥16 and required ventilator support for more than 48 h in the intensive care unit. Next-generation sequencing was used to profile the miRNAs and mRNAs expressed in CD3+ T cells isolated from patient blood samples collected during the injury and recovery stages.
Results
In the 26 studied patients, 9 miRNAs (hsa-miR-16-2-3p, hsa-miR-16-5p, hsa-miR-185-5p, hsa-miR-192-5p, hsa-miR-197-3p, hsa-miR-23a-3p, hsa-miR-26b-5p, hsa-miR-223-3p, and hsa-miR-485-5p) were significantly upregulated, while 58 mRNAs were significantly downregulated in T cells following major trauma. A network consisting of 8 miRNAs and 22 mRNAs interactions was revealed by miRWalk, with three miRNAs (hsa-miR-185-5p, hsa-miR-197-3p, and hsa-miR-485-5p) acting as hub genes that regulate the network. Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis suggested that “chemokine signaling pathway” was the predominant pathway.
Conclusion
The study revealed a miRNA-mRNA interactome consisting of 8 miRNAs and 22 mRNAs that are predominantly involved in chemokine signaling in circulating T cells of patients following major trauma.
Introduction
Major trauma can lead to a systemic inflammatory response and disrupted immune system homeostasis.Citation1,Citation2 The cytokine/genomic “storm” that occurs after major trauma is well describedCitation3,Citation4 and involves the simultaneous upregulation of genes involved in systemic inflammatory responses and the downregulation of genes involved in adaptive immunity.Citation3 These immunoinflammatory responses greatly impact the prognosis of patients with major trauma.Citation5 Prolonged immune suppression may lead to increased susceptibility to secondary infections.Citation7 A retrospective analysis of 917 patients with an injury severity score (ISS) ≥16 revealed that the absolute lymphocyte counts on day 3 and the ratio of absolute lymphocyte counts on day 3 and day 1 after trauma are independent risk factors for sepsis and patient mortality.Citation8 In addition, the extent of immune dysfunction following major trauma is strongly correlated to patient morbidity and mortality.Citation6
Immunosuppression after major trauma is characterized primarily using T cell populations of the adaptive immune system.Citation7,Citation9 Weighted gene co-expression network analysis of trauma patients revealed that modules with high T cell activation and low neutrophil activation indicate better survival of patients with sepsis.Citation10 Genome-wide expression analysis of whole-blood leukocytes in 167 adult patients with severe blunt trauma revealed widespread transcriptome changes, with alterations in up to 80% of the leukocyte transcriptome.Citation3 The genomic response to traumatic injury involves the upregulation of several genes involved in the mediation of inflammation, pattern recognition, and antimicrobial activity, along with downregulation of genes engaged in antigen presentation, natural killer (NK) cell function, and T cell proliferation and apoptosis.Citation3 Of the interleukin (IL)-10, IL-6, and p38 mitogen-activated protein kinase (MAPK) pathways are the most upregulated pathways associated with complicated recovery, whereas T cell regulation and antigen presentation are among the most downregulated pathways.Citation3
MicroRNAs (miRNAs) are endogenous small non-coding RNAs that regulate gene expression through post-transcriptional modulation,Citation11 primarily by binding to the 3′ UTR of target mRNAs, which leads to mRNA degradation or translation inhibition.Citation12 miRNAs are important regulators of almost all cellular processes,Citation13 including the immune system.Citation14–16 The loss of certain miRNAs or genetic ablation of miRNA machinery can severely compromise immune system development and immune responses, thus leading to immune dysfunction.Citation14 A high-throughput study identified 69 dysregulated circulating miRNAs in patients with major trauma 12 h after intensive care unit (ICU) admission compared to healthy controls.Citation17 Among these dysregulated miRNAs, 14 were correlated with innate immune responses involving the toll-like receptor (TLR) 3, TLR4, myeloid differentiation primary response 88 (MYD88), and Toll-receptor-associated molecule (TRAM) pathways. However, little is known about the role of miRNAs in major trauma, especially in T cells of the adaptive immune system. Therefore, we investigated dysregulated miRNAs and potential mRNA targets in circulating T cells of major trauma patients. To reduce patient-to-patient variations, we used samples from the same patients during the recovery stage as control samples.
Patients and Methods
Patient Enrollment
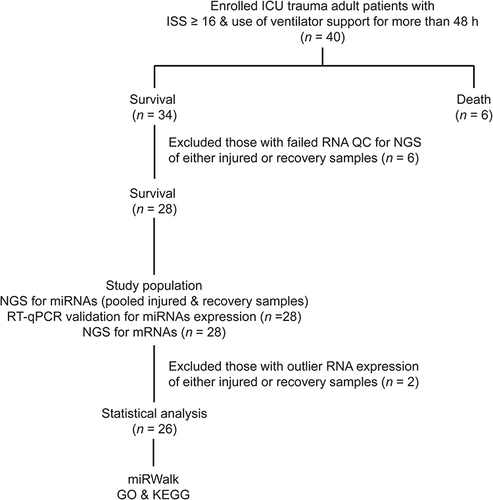
Trauma patients were recruited as study participants after admission to the trauma ICU of a level I trauma center in Southern TaiwanCitation18–20 between December 2017 and December 2018. Only patients who fit the following criteria were included: (1) age of 20 years or above; (2) admission to the ICU due to trauma injury; (3) ISS ≥ 16;Citation21–23 and (4) the use of ventilator support for more than 48 h. Patients who fit the following criteria were excluded: (1) immunocompromised condition; (2) malignancy; or (3) unwillingness to participate in the study. Forty trauma patients with critical illness who stayed in the ICU were enrolled in the study (). Written informed consent was collected from each participant. Patient medical information, including sex, age, Glasgow Coma Scale (GCS), Abbreviated Injury Scale (AIS) in each body region, and ISS, was collected. The AIS is the basis of many severity scoring systems that assess the severity of an anatomical injury on a six-point ordinal scale ranging from 1 to 6 as minor, moderate, serious, severe, critical, and un-survivable, respectively.
Figure 1 Enrollment of the patients and flowchart illustrating the collection and processing of samples for the experiments.
Specimen Collection
Peripheral blood samples were collected from each patient within 72 h after admission to the ICU. Fasting blood samples (10 mL) were collected from the forearm at this time point and were defined injury samples. After the patients successfully left the ICU, another peripheral blood sample was collected immediately before discharge from patients who stayed in the hospital. The blood samples drawn at this time point were defined recovery samples. If the patients did not survive, collection of the second blood sample could not be performed. This study only compared miRNA and mRNA expression in injury and recovery samples from the same patients. Vacutainer Serum Separator Tubes (BD Diagnostics, Franklin Lakes, NJ, USA) were used to obtain serum from the blood samples without using additives. The collected blood samples were preserved in ice and were immediately sent to the laboratory. Ficoll–Paque Premium (17–5442-02, Merck, Kenilworth, NJ, USA) was used to separate the peripheral blood mononuclear cells (PBMCs) from whole blood by the density gradient centrifugation. Four-milliliter Ficoll solution was added to the bottom of a 15 mL tube before adding 8 mL whole blood. PBMCs were collected from the Ficoll/plasma interface after centrifugation at 400 g for 40 min at 25 ℃. The samples were diluted by mixing equal amounts of phosphate-buffered saline. After centrifuging the diluted samples at 240 g for 5 min, the supernatant was removed and the cell pellets were resuspended in 1X BD IMag buffer (BD Diagnostics). Cell numbers were counted. The cell suspensions were centrifuged at 200 g for 10 min. For every 107 cells in the supernatant, 50 μL of BD IMag anti-human CD3 magnetic particles (552,593, BD Bioscience) was added. The mixtures were then incubated at 37 ℃ for 30 min. After washing with an equal amount of BD IMag buffer, CD3+ cell pellets were collected and resuspended in 500 μL QIAzol lysis reagent from an RNeasy Mini Kit (74,104, Qiagen, Venlo, Netherlands). RNeasy Mini Kits (Qiagen) was used to extract total RNA, which was subsequently quantified using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA) and a Qubit RNA Assay Kit (Q10210, Thermo Scientific). A Caliper LabChip Analyzer (PerkinElmer, Waltham, MA, USA) was used to determine the RNA quality based on the RNA integrity number (RIN).
Next-Generation Sequencing (NGS) Analysis of miRNAs
Total RNA from the injury samples (n = 10) and the corresponding recovery samples (n = 10) were pooled for every five patients to create two pooled injury samples (n = 2) and two pooled recovery samples (n = 2). GeneTech Biotech Co., Ltd (GeneTech, Taipei, Taiwan) performed the small RNA cloning and NGS analysis. Briefly, miRNA 15–30 nucleotides long were passively eluted from polyacrylamide gel, precipitated with ethanol and melted in double-distilled water. An Illumina TruSeq Small RNA Sample Prep Kit was used to ligate small RNA linkers onto prepared bar-coded cDNAs. 3′ and 5′ adapter-ligated RNA in 1 μg total RNA was reverse-transcribed with Invitrogen SuperScript II Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA) and was then amplified by 15 cycles of polymerase chain reaction (PCR). To reduce sample bias, 15 barcoded reverse primers were ligated directly to the miRNAs. A BioAnalyzer 2100 (Agilent Technologies) was used to analyze individual libraries to evaluate the presence of linked cDNA and 15 bar-coded libraries (135–165 bp).
RT-qPCR for miRNA Expression
The expression of significantly upregulated miRNAs identified by NGS analysis of the injury and recovery samples (n = 28 each) was determined by real-time quantitative reverse transcription polymerase chain reaction (RT-qPCR). TaqMan MicroRNA Reverse Transcription Kits (Applied Biosystems, Foster City, CA, USA) were used to reverse transcribe the RNA into cDNA. Then, TaqMan Universal PCR Master Mix (No UNG, PN 4324018, Applied Biosystems) and specific miRNA primers from TaqMan MicroRNA Assays (Applied Biosystems) were used to amplify the cDNA. About 25 fmol single-stranded cel-miR-39 (Invitrogen) was added to each sample for use as an internal control to determine miRNA expression. RT-qPCR was performed using a 7500 RT-qPCR system (Applied Biosystems) and SYBR Green (Applied Biosystems). miRNA expression levels were quantified using the 2−ΔΔCt method using normalized cycle threshold (Ct) values. Beta-actin was used as the internal control. When the mean value of all the samples was different from that of its control by more than two-fold and p < 0.05, the change in miRNA expression was considered statistically significant.
Next-Generation Sequencing of mRNAs
mRNA expression in the injury and recovery samples (n = 28, each) was determined by GeneTech Biotech Co., Ltd (GeneTech). NGS libraries were constructed using a NEBNext Ultra Directional RNA Library Prep Kit (Illumina). The rRNA was removed from each sample using a Ribo-Zero rRNA removal Kit (Illumina). Then, the rRNA-depleted RNA was fragmented and reverse-transcribed. ProtoScript II Reverse Transcriptase with random primers and actinomycin-D (Illumina) and Second Strand Synthesis Enzyme Mix (Illumina) were used to synthesize the first- and second-strand cDNA, respectively. An AxyPrep Mag PCR Clean-up kit (Axygen, New York, NY, USA) was used to purify the double-stranded cDNA. End Prep Enzyme Mix (Axygen) was used to repair both ends, add a dA-tail, and ligate adaptors to both ends of the cDNA fragments. An AxyPrep Mag PCR Clean-up kit (Axygen) was used to select the size of adaptor-ligated DNA (~360 bp). Uracil-Specific Excision Reagent enzyme (New England Biolabs, Ipswich, MA, USA) was used to digest the dUTP-marked second-strand. Each sample was amplified for 11 PCR cycles using P5 and P7 primers. Libraries were sequenced using a HiSeq 2000 sequencing system (Illumina) using paired-end reads. HiSeq Control Software and OLB with GAPipeline-1.6.0 (Illumina) were used for image analysis and base calling. GENEWIZ (South Plainfield, NJ, USA) processed and analyzed the sequencing data. Trimmomatic v0.30 was used to filter the data in FASTQ format to get high-quality clean data. Fragment counts were determined using Hisat2 v2.0.1 and human reference genome sequences hg19 obtained from the UCSC website. Gene and isoform expression levels were determined from cleaned data using HTSeq v0.6.1.
Analysis of miRNA–mRNA Interactions and Their Functions
miRWalk version 2.0 software (http://mirwalk.uni-hd.de),Citation24 a publicly available comprehensive resource for miRNA–mRNA interaction pairs, was used to explore the miRNA-mRNA interactome. The miRWalk hosts possible binding site interaction information between miRNAs and target mRNAs based on TarPmiR (http://hulab.ucf.edu/research/projects/miRNA/TarPmiR),Citation25 miRNA-target prediction data from TargetScan (https://www.targetscan.org)Citation26 and miRDB (http://www.mirdb.org),Citation27 and validated interaction data from miRTarBase (https://mirtarbase.cuhk.edu.cn).Citation28 All genes were input to the miRWalk website and analyzed using the default settings to produce miRNA–gene interaction output tables. Cytoscape version 3.40 (https://cytoscape.org) was used to visualize the miRNA-mRNA interactome. To explore the function of the mRNA targets identified in the miRNA-mRNA interactome, gene ontology (GO) enrichment analysisCitation29 was performed using the DAVID 2021 database (https://david.ncifcrf.gov/). The KEGG Orthology-Based Annotation System 3.0 (http://kobas.cbi.pku.edu.cn/)Citation30 was used to analyze and illustrate pathways from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database.
Statistical Analysis
R 3.3.3 software (The R foundation) was used to process and analyze the data. Differential expression analysis of miRNAs and mRNAs between the injury and recovery samples was performed using edgeR package (ver 3.15). Benjamini and Hochberg’s approach was performed to control the false discovery rate. miRNA expression from RT-qPCR was analyzed using non-parametric Mann–Whitney U-tests using the Wilcox. test function in R. Statistical significance was indicated by two-sided p < 0.05.
Results
Patient Characteristics
Of the 40 enrolled patients, six died and 34 survived (). After excluding patients with failed RNA-seq quality control in either the injury or recovery samples (n = 6), 28 pairs of injured and recovery samples were used to quantify miRNA expression (by RT-qPCR) and mRNA (NGS). After excluding two patients with outlier RNA expression (defined as a data point having less than Q1 - 1.5 × interquartile range (IQR) or higher than Q3 – 1.5 × IQR) in either the injury or recovery samples, 26 patients with both injury and recovery samples were selected for further analysis. Among these 26 patients (), 14 were male and 12 were female with an average age of 55.8 ± 17.6 years (minimum 22 years and maximum 64 years). Based on injury severity (AIS ≥ 2), the head was the most frequently injured body region (80.8%), followed by the extremities (46.1%), thorax (42.3%), and abdomen (23.0%). The median consciousness level according to the GCS was 12 (Q1-Q3, 6–15) and the median ISS was 20 (Q1-Q3, 16–25). The time between the collection of injury samples and recovery samples was 24.2 ± 7.2 days.
Table 1 The Patient and Injury Characteristics of the Enrolled and Studied Cohorts
miRNA and mRNA Expression
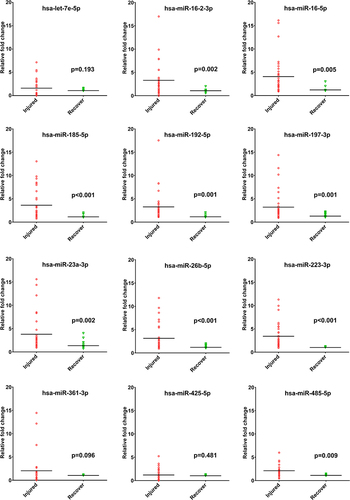
NGS analysis of miRNAs revealed that twelve miRNAs (hsa-let-7e-5p, hsa-miR-16-2-3p, hsa-miR-16-5p, hsa-miR-185-5p, hsa-miR-192-5p, hsa-miR-197-3p, hsa-miR-23a-3p, hsa-miR-26b-5p, hsa-miR-223-3p, hsa-miR-361-3p, hsa-miR-425-5p, and hsa-miR-485-5p) were more abundant in the circulating T cells of patients in the injury stage, compared to those in the recovery stage. As shown in and , RT-qPCR revealed 9 significantly upregulated miRNAs (hsa-miR-16-2-3p, hsa-miR-16-5p, hsa-miR-185-5p, hsa-miR-192-5p, hsa-miR-197-3p, hsa-miR-23a-3p, hsa-miR-26b-5p, hsa-miR-223-3p, and hsa-miR-485-5p) in the injury samples than the recovery samples. No miRNAs were downregulated in the injury samples compared to the recovery samples. In addition, 58 mRNAs were significantly downregulated in circulating T cells of patients in the injury stage compared to patients in the recovery stage ().
Table 2 The Up-Regulated miRNAs Identified from the Next-Generation Sequencing in the Injury and Recovery Samples. The Fold Expression of miRNAs Was Expressed as Mean (Standard Deviation)
Table 3 Down-Regulated mRNA Targets Identified from the Next-Generation Sequencing Analysis in the Injury Samples Compared to Those in the Recovery Samples
Figure 2 The real-time quantitative reverse transcription polymerase chain reaction (RT-qPCR) to determine the expression of the 12 up-regulated miRNAs, which were identified from the next-generation sequencing analysis in the injury samples compared to those in the recovery samples.
miRNA–mRNA Interactions and Functions
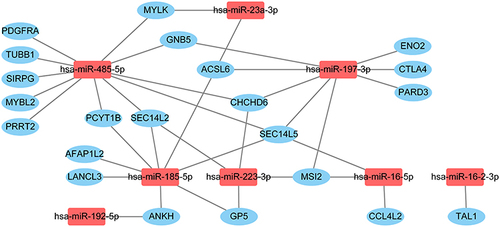
GO enrichment analysis of the 58 mRNAs revealed 6 biological processes, 2 cellular components, and 1 molecular function (). In the biological process category, “positive regulation of synapse assembly” and “cell adhesion” were the two most enriched terms in the respective categories. For cellular components, “plasma membrane” and “extracellular space” were the most enriched terms, while “transmembrane receptor protein tyrosine kinase activator activity” was the most enriched molecular function term. To explore miRNA–mRNA interactions, we analyzed the 9 upregulated miRNAs and the 58 downregulated mRNAs using miRWalk. We constructed a node graph of the interactions between the 8 miRNAs and 22 mRNAs (). Three miRNAs (hsa-miR-185-5p, hsa-miR-197-3p, and hsa-miR-485-5p) were identified as hub miRNAs (defined by connections with more than three mRNAs) that regulate the miRNA-mRNA interactome. There were no connections between hsa-miR-26b-5p and any of the 58 mRNA targets. GO analysis of the 22 mRNAs involved in the miRNA–mRNA interactions indicated that “plasma membrane” (cellular components) was the only enriched term (). KEGG pathway enrichment analysis suggested that the “chemokine signaling pathway” (pathway ID: hsa04062) was the predominant pathway. Three genes (PARD3, CCL4L2, GNB5) were involved in the pathway (p = 3.6E-04).
Table 4 Gene Ontology Enrichment Analysis of 58 Differentially Expressed mRNAs Between the Samples of Patients in the Injured Stage Vs Recovery Stage
Table 5 The Enrichment Analysis of the Gene Ontology (GO) Terms of the Down-Regulated mRNAs in the Constructed miRNA-mRNA Interactome. mRNA Interaction
Figure 3 The node graph of the interactions between 8 miRNAs and 22 mRNAs identified from miRWalk.
Discussion
The present study indicates that following major trauma, nine miRNAs were significantly upregulated in the circulating T cells of patients in the injury stage compared to those of the same patients in the recovery stage. The miRNA-mRNA interactome consisted of 8 miRNAs and 22 mRNAs and was used to analyze the functions of the involved genes. The GO term, “plasma membrane”, and the KEGG pathway term, “chemokine signaling pathway”, were predominant.
Chemokines are small chemoattractant peptides that provide directional cues for cell trafficking and thus play a pivotal role in host response protection,Citation31,Citation32 inflammatory responses, immune homeostasis, and cancer progression.Citation33 Some chemokines are induced during an immune response to recruit immune cells to infection sites and are thus considered pro-inflammatory, while some chemokines are considered to be homeostatic and are involved in regulating cell migration during development or tissue maintenance.Citation31,Citation32 Impaired chemokine signaling pathway in T cells controlled by miRNA–mRNA interactions may partly explain the immunosuppression in patients following major trauma. Xiao et al found that even after different injuries, there is an apparently fundamental response to severe inflammatory stress characterized by common genomic signatures.Citation3 Regarding the most significantly regulated pathways, injury led to early activation of innate immune responses and simultaneous suppression of adaptive immune responses. Injury severity, clinical outcomes, magnitude of physiological decline, and transfused blood volume only minimally affected these patterns.Citation3 Interestingly, some studies demonstrated activation of chemokine signaling pathway genes in sciatic nerve injury,Citation34 traumatic brain injury,Citation35 and acute and chronic spinal cord injuries.Citation36,Citation37 Notably, most patients (16 of 26 patients) in this study had traumatic brain injuries. The impact of injuries to various body parts on specific miRNA–mRNA interactions requires further investigation.
Three miRNAs (hsa-miR-185-5p, hsa-miR-197-3p, and hsa-miR-485-5p) were identified as hub miRNAs that regulate the miRNA–mRNA interactions. Hsa-miR-185-5p is involved in the development of Alzheimer’s diseaseCitation38,Citation39 and acquired chemotherapy resistance in patients with gastric cancer.Citation40 Overexpression of hsa-miR-185-5p occurs in the vitreous of proliferative diabetic retinopathy patients,Citation41 in the pericardiac adipose tissue of patients with coronary artery disease,Citation42 and significantly enhances endothelial cell angiogenesis.Citation43 hsa-miR-197-3p is highly expressed in proliferative diabetic retinopathy patients,Citation44,Citation45 is involved in the development of colorectal cancerCitation46 and non-small cell lung cancer,Citation47 and may act as a biomarker for follicular thyroid cancer.Citation48 Further, hsa-miR-485-5p is associated with lupus nephritis,Citation49 pulmonary tuberculosis,Citation50 and cancers such as lung cancer,Citation51 hepatocellular carcinoma,Citation52 oral tongue squamous cell carcinoma,Citation53 and glioblastoma.Citation54 None of these three miRNAs have so far been reported to be associated with T cell function or adaptive immunity. Therefore, investigating the impact of these miRNA–mRNA interactions on immune functions may be necessary to understand immune dysfunction in patients with major trauma.
This study has some limitations. First, a small, single-institution patient population was used to study a complex network of miRNA–mRNA interactions. Second, some factors such as damage control, resuscitation, surgery, and drug use may lead to bias. Third, the expression of these circulating molecules may be dynamic in nature and conclusions drawn from two single measurements may not reflect changes over time. Further, although the study population consisted of severe trauma patients with an ISS ≥ 16 and requiring more than 48 h ventilator support, these patients sustained injuries to different body parts, potentially leading to selection bias in the study. However, we believe that identifying the miRNA–mRNA interactions may provide valuable information for understanding immune dysfunction in patients with major trauma. Finally, the results derived from this study depend on which trauma is the predominant injury type, considering that patients with traumatic brain injuries comprised most of the study population.
Conclusion
Our results reveal that, following major trauma, nine miRNAs are significantly upregulated in the circulating T cells of trauma patients in the injury stage compared to those of the same patients in the recovery stage. A miRNA-mRNA interactome consisting of 8 miRNAs and 22 mRNAs is involved in regulating the chemokine signaling pathway after major trauma.
Ethics Approval and Informed Consent
The study was approved by the Institutional Review Board of Chang Gung Memorial Hospital (ref: 201600221B0 and 201701425B0) under the ethical guidelines of the 1975 Declaration of Helsinki. Written informed consent was obtained from all subjects involved in the study.
Disclosure
The authors report no conflicts of interest in this work.
Acknowledgments
We are grateful for support provided by the Biostatistics Center of the Kaohsiung Chang Gung Memorial Hospital.
Additional information
Funding
References
- Di Battista AP, Rhind SG, Hutchison MG, et al. Inflammatory cytokine and chemokine profiles are associated with patient outcome and the hyperadrenergic state following acute brain injury. J Neuroinflammation. 2016;13:40. doi:10.1186/s12974-016-0500-3
- Catania A, Lonati C, Sordi A, Gatti S. Detrimental consequences of brain injury on peripheral cells. Brain Behav Immun. 2009;23:877–884. doi:10.1016/j.bbi.2009.04.006
- Xiao W, Mindrinos MN, Seok J, et al. A genomic storm in critically injured humans. J Exp Med. 2011;208:2581–2590. doi:10.1084/jem.20111354
- Wu SC, Rau CS, Kuo PJ, et al. Profiling the expression of circulating acute-phase proteins, cytokines, and checkpoint proteins in patients with major trauma: a pilot study. J Inflamm Res. 2021;14:3739–3753. doi:10.2147/jir.S324056
- Bonaroti J, Abdelhamid S, Kar U, et al. The use of multiplexing to identify cytokine and chemokine networks in the immune-inflammatory response to trauma. Antioxid Redox Signal. 2021;35:1393–1406. doi:10.1089/ars.2021.0054
- Finlay LD, Conway Morris A, Deane AM, Wood AJ. Neutrophil kinetics and function after major trauma: a systematic review. World J Crit Care Med. 2021;10:260–277. doi:10.5492/wjccm.v10.i5.260
- Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–150. doi:10.1056/NEJMra021333
- Dong X, Wang C, Liu X, Bai X, Li Z. The trajectory of alterations in immune-cell counts in severe-trauma patients is related to the later occurrence of sepsis and mortality: retrospective study of 917 cases. Front Immunol. 2020;11:603353. doi:10.3389/fimmu.2020.603353
- Keel M, Trentz O. Pathophysiology of polytrauma. Injury. 2005;36:691–709. doi:10.1016/j.injury.2004.12.037
- Huo J, Wang L, Tian Y, et al. Gene co-expression analysis identified preserved and survival-related modules in severe blunt trauma, burns, sepsis, and systemic inflammatory response syndrome. Int J Gen Med. 2021;14:7065–7076. doi:10.2147/ijgm.S336785
- Vidigal JA, Ventura A. The biological functions of miRNAs: lessons from in vivo studies. Trends Cell Biol. 2015;25:137–147. doi:10.1016/j.tcb.2014.11.004
- O’Brien J, Hayder H, Zayed Y, Peng C. Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol. 2018;9:402. doi:10.3389/fendo.2018.00402
- Hirschberger S, Hinske LC, Kreth S. MiRNAs: dynamic regulators of immune cell functions in inflammation and cancer. Cancer Lett. 2018;431:11–21. doi:10.1016/j.canlet.2018.05.020
- Xiao C, Rajewsky K. MicroRNA control in the immune system: basic principles. Cell. 2009;136:26–36. doi:10.1016/j.cell.2008.12.027
- Nanbakhsh A, Malarkannan S. The role of microRNAs in NK cell development and function. Cells. 2021;10:2020. doi:10.3390/cells10082020
- Xiao C, Nemazee D, Gonzalez-Martin A. MicroRNA control of B cell tolerance, autoimmunity and cancer. Semin Cancer Biol. 2020;64:102–107. doi:10.1016/j.semcancer.2019.04.004
- Uhlich RM, Konie JA, Davis JW, et al. Novel microRNA correlations in the severely injured. Surgery. 2014;156:834–840. doi:10.1016/j.surg.2014.06.017
- Hsieh CH, Hsu SY, Hsieh HY, Chen YC. Differences between the sexes in motorcycle-related injuries and fatalities at a Taiwanese level I trauma center. Biomed J. 2017;40:113–120. doi:10.1016/j.bj.2016.10.005
- Hsieh CH, Liu HT, Hsu SY, Hsieh HY, Chen YC. Motorcycle-related hospitalizations of the elderly. Biomed J. 2017;40:121–128. doi:10.1016/j.bj.2016.10.006
- Hsieh CH, Chen YC, Hsu SY, Hsieh HY, Chien PC. Defining polytrauma by abbreviated injury scale >/= 3 for a least two body regions is insufficient in terms of short-term outcome: a cross-sectional study at a level I trauma center. Biomed J. 2018;41:321–327. doi:10.1016/j.bj.2018.08.007
- Hsu SY, Wu SC, Rau CS, et al. Impact of adapting the Abbreviated Injury Scale (AIS)-2005 from AIS-1998 on injury severity scores and clinical outcome. Int J Environ Res Public Health. 2019;16:5033. doi:10.3390/ijerph16245033
- Stewart KE, Cowan LD, Thompson DM. Changing to AIS 2005 and agreement of injury severity scores in a trauma registry with scores based on manual chart review. Injury. 2011;42:934–939. doi:10.1016/j.injury.2010.05.033
- Newgard CD, Fu R, Lerner EB, et al. Deaths and high-risk trauma patients missed by standard trauma data sources. J Trauma Acute Care Surg. 2017;83:427–437. doi:10.1097/ta.0000000000001616
- Sticht C, De La Torre C, Parveen A, Gretz N. miRWalk: an online resource for prediction of microRNA binding sites. PLoS One. 2018;13:e0206239. doi:10.1371/journal.pone.0206239
- Ding J, Li X, Hu H. TarPmiR: a new approach for microRNA target site prediction. Bioinformatics. 2016;32:2768–2775. doi:10.1093/bioinformatics/btw318
- McGeary SE, Lin KS, Shi CY, et al. The biochemical basis of microRNA targeting efficacy. Science. 2019;366. doi:10.1126/science.aav1741
- Chen Y, Wang X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020;48:D127–d131. doi:10.1093/nar/gkz757
- Huang HY, Lin YC, Li J, et al. miRTarBase 2020: updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res. 2020;48:D148–d154. doi:10.1093/nar/gkz896
- Gene Ontology Consortium. The Gene Ontology (GO) project in 2006. Nucleic Acids Res. 2006;34:D322–326. doi:10.1093/nar/gkj021
- Draghici S, Khatri P, Tarca AL, et al. A systems biology approach for pathway level analysis. Genome Res. 2007;17:1537–1545. doi:10.1101/gr.6202607
- Rajarathnam K, Schnoor M, Richardson RM, Rajagopal S. How do chemokines navigate neutrophils to the target site: dissecting the structural mechanisms and signaling pathways. Cell Signal. 2019;54:69–80. doi:10.1016/j.cellsig.2018.11.004
- Zaja-Milatovic S, Richmond A. CXC chemokines and their receptors: a case for a significant biological role in cutaneous wound healing. Histol Histopathol. 2008;23:1399–1407. doi:10.14670/hh-23.1399
- Xun Y, Yang H, Li J, Wu F, Liu F. CXC chemokine receptors in the tumor microenvironment and an update of antagonist development. Rev Physiol Biochem Pharmacol. 2020;178:1–40. doi:10.1007/112_2020_35
- Li L, Du X, Ling H, et al. Gene correlation network analysis to identify regulatory factors in sciatic nerve injury. J Orthop Surg Res. 2021;16:622. doi:10.1186/s13018-021-02756-0
- Cao XY, Qian X, Liu GD, Wang YH. Bioinformatics-based identification of key pathways and hub genes of traumatic brain injury in a rat model. Curr Med Sci. 2021;41:610–617. doi:10.1007/s11596-021-2365-7
- Niu SP, Zhang YJ, Han N, Yin XF, Zhang DY, Kou YH. Identification of four differentially expressed genes associated with acute and chronic spinal cord injury based on bioinformatics data. Neural Regen Res. 2021;16:865–870. doi:10.4103/1673-5374.297087
- Zhu Z, Shen Q, Zhu L, Wei X. Identification of pivotal genes and pathways for spinal cord injury via bioinformatics analysis. Mol Med Rep. 2017;16:3929–3937. doi:10.3892/mmr.2017.7060
- Sabaie H, Talebi M, Gharesouarn J, et al. Identification and analysis of BCAS4/hsa-miR-185-5p/SHISA7 competing endogenous RNA axis in late-onset alzheimer’s disease using bioinformatic and experimental approaches. Front Aging Neurosci. 2022;14:812169. doi:10.3389/fnagi.2022.812169
- Roy J, Mallick B. Altered gene expression in late-onset Alzheimer’s disease due to SNPs within 3’UTR microRNA response elements. Genomics. 2017;109:177–185. doi:10.1016/j.ygeno.2017.02.006
- Sun J, Zhao J, Yang Z, Zhou Z, Lu P. Identification of gene signatures and potential therapeutic targets for acquired chemotherapy resistance in gastric cancer patients. J Gastrointest Oncol. 2021;12:407–422. doi:10.21037/jgo-21-81
- Friedrich J, Steel DHW, Schlingemann RO, et al. microRNA expression profile in the vitreous of proliferative diabetic retinopathy patients and differences from patients treated with Anti-VEGF therapy. Transl Vis Sci Technol. 2020;9:16. doi:10.1167/tvst.9.6.16
- Li M, Qi L, Li Y, et al. Association of pericardiac adipose tissue with coronary artery disease. Front Endocrinol. 2021;12:724859. doi:10.3389/fendo.2021.724859
- Zhong XQ, Yan Q, Chen ZG, et al. Umbilical cord blood-derived exosomes from very preterm infants with bronchopulmonary dysplasia impaired endothelial angiogenesis: roles of exosomal MicroRNAs. Front Cell Dev Biol. 2021;9:637248. doi:10.3389/fcell.2021.637248
- Guo J, Zhou P, Liu Z, et al. The aflibercept-induced MicroRNA profile in the vitreous of proliferative diabetic retinopathy patients detected by next-generation sequencing. Front Pharmacol. 2021;12:781276. doi:10.3389/fphar.2021.781276
- Guo J, Zhou P, Pan M, et al. Relationship between elevated microRNAs and growth factors levels in the vitreous of patients with proliferative diabetic retinopathy. J Diabetes Complications. 2021;35:108021. doi:10.1016/j.jdiacomp.2021.108021
- Liu N, Jiang F, Chen Z. A preliminary study on the pathogenesis of colorectal cancer by constructing a Hsa-circRNA-0067835-miRNA-mRNA regulatory network. Onco Targets Ther. 2021;14:4645–4658. doi:10.2147/ott.S319300
- Zhang Q, Kang L, Li X, Li Z, Wen S, Fu X. Bioinformatics analysis predicts hsa_circ_0026337/miR-197-3p as a potential oncogenic ceRNA network for non-small cell lung cancers. Anticancer Agents Med Chem. 2022;22:874–886. doi:10.2174/1871520621666210712090721
- Stokowy T, Eszlinger M, Świerniak M, et al. Analysis options for high-throughput sequencing in miRNA expression profiling. BMC Res Notes. 2014;7:144. doi:10.1186/1756-0500-7-144
- Navarro-Quiroz E, Pacheco-Lugo L, Navarro-Quiroz R, et al. Profiling analysis of circulating microRNA in peripheral blood of patients with class IV lupus nephritis. PLoS One. 2017;12:e0187973. doi:10.1371/journal.pone.0187973
- Li ZB, Shi LY, Han YS, et al. Pyridoxal phosphate, pyridoxamine phosphate, and folic acid based on ceRNA regulatory network as potential biomarkers for the diagnosis of pulmonary tuberculosis. Infect Genet Evol. 2022;99:105240. doi:10.1016/j.meegid.2022.105240
- Jiang P, Xu C, Chen L, et al. Epigallocatechin-3-gallate inhibited cancer stem cell-like properties by targeting hsa-mir-485-5p/RXRα in lung cancer. J Cell Biochem. 2018;119:8623–8635. doi:10.1002/jcb.27117
- Peng Y, Leng W, Duan S, Hong M. Long noncoding RNA BLACAT1 is overexpressed in hepatocellular carcinoma and its downregulation suppressed cancer cell development through endogenously competing against hsa-miR-485-5p. Biomed Pharmacother. 2019;116:109027. doi:10.1016/j.biopha.2019.109027
- Lin XJ, He CL, Sun T, Duan XJ, Sun Y, Xiong SJ. hsa-miR-485-5p reverses epithelial to mesenchymal transition and promotes cisplatin-induced cell death by targeting PAK1 in oral tongue squamous cell carcinoma. Int J Mol Med. 2017;40:83–89. doi:10.3892/ijmm.2017.2992
- Basso J, Paggi MG, Fortuna A, Vitorino C, Vitorino R. Deciphering specific miRNAs in brain tumors: a 5-miRNA signature in glioblastoma. Mol Genet Genomics. 2022;297:507–521. doi:10.1007/s00438-022-01866-6