Abstract
Background
Idiopathic pulmonary fibrosis (IPF) is a fatal lung disease with no known effective therapy. It is often assumed, but has not been objectively evaluated, that pulmonary inflammation subsides as IPF progresses. The goal of this work was to assess changes in the degree of inflammatory cell infiltration, particularly lymphocytic infiltration, over the duration of illness in IPF.
Methods
Sixteen patients with confirmed IPF were identified in patients whom surgical lung biopsy (SLB) was performed in early disease, and in patients whom lung transplantation was subsequently performed in end stage disease. A numerical scoring system was used to histologically quantify the amount of fibrosis, honeycomb change, fibroblastic foci, and lymphocyte aggregates in each SLB and lung explant tissue sample. Analyses of quantitative scores were performed by comparing paired, matched samples of SLB to lung explant tissue.
Results
Median time [1st, 3rd quartiles] from SLB to lung transplantation was 24 [15, 29] months. Histologic fibrosis and honeycomb change were more pronounced in the explant samples compared with SLB (P < 0.001 and P < 0.01, respectively), and most notably, higher numbers of lymphocyte aggregates were observed in the explant samples compared to SLB (P = 0.013). Immunohistochemical analyses revealed abundant CD3+ (T lymphocyte) and CD20+ (B lymphocyte) cells, but not CD68+ (macrophage) cells, within the aggregates.
Conclusion
Contrary to the frequent assumption, lymphocyte aggregates were present in greater numbers in advanced disease (explant tissue) compared to early disease (surgical lung biopsy). This finding suggests that active cellular inflammation continues in IPF even in severe end stage disease.
Introduction
Inflammation in the lung is a consistent finding in patients with idiopathic interstitial pneumonia, a group of pulmonary diseases characterized by distinct clinical, physiologic, radiologic, and pathologic findings.Citation1 The most common and severe form of idiopathic interstitial pneumonia is idiopathic pulmonary fibrosis (IPF). The pathologic hallmark of IPF is a pattern of lung injury termed usual interstitial pneumonitis (UIP), which consists histologically of a combination of inflammatory cell infiltration, loss of normal epithelium, clusters of fibroblasts or myofibroblasts (fibroblastic foci), extracellular matrix deposition (fibrosis), and the formation of small lung cysts (honeycomb change).Citation2,Citation3 IPF is a severely debilitating and fatal lung disease, with an overall median survival of approximately 3 years, with no proven effective therapy, and with lung transplantation remaining the only viable intervention in end stage disease.Citation3
Although the precise role of inflammation in IPF remains controversial, there appears to be little doubt that inflammation is consistently present in the lungs of these patients. From the earliest descriptions of patients with pulmonary fibrosis, infiltration of inflammatory cells in the lung parenchyma, particularly lymphocytes, was a universally observed histologic finding,Citation4,Citation5 and almost all patients currently diagnosed with IPF will manifest a mild to moderate degree of inflammatory cell infiltration in the lung.Citation1–Citation3,Citation6,Citation7 In addition to the presence of inflammatory cells in IPF, increased levels of numerous pro-inflammatory cytokines, chemokines, and cell surface molecules have been extensively documented over the last four decades in animal models and patients with pulmonary fibrosis.Citation8–Citation11
It has been suggested that patients with IPF have active inflammatory cell infiltration early in the course of disease, but subsequently have decreasing amounts of inflammation as the disease progresses over time, as fibrosis ensues, and as the inflammatory process becomes “burned out.”Citation5,Citation12–Citation14 This concept of inflammation representing an early stage of IPF and disappearing at later stages of disease may seem plausible, but the degree of inflammatory cell infiltration at different time points in IPF has never been objectively evaluated.
The goal of the current study was to histologically assess the degree of inflammatory cell infiltration as IPF progresses. We identified a cohort of 16 patients with IPF in whom a surgical lung biopsy (SLB) was performed as part of the initial diagnostic evaluation (early disease) and in whom lung transplantation (with available lung explant tissue) was performed at a later time point due to the development of end stage lung disease. By performing a detailed qualitative and quantitative histologic assessment on each SLB and explant tissue sample, we sought to determine the degree of inflammatory cell infiltration, particularly lymphocytic cellular infiltration, in IPF over the duration of patient disease.
Patients and methods
Patients
This study was reviewed and approved by the University of Maryland Institutional Review Board. One hundred fifty seven patients underwent lung transplantation at the University of Maryland from 2002 through 2011, and from this group, we identified 16 patients with IPF in whom a SLB was performed early in their illness as part of the initial diagnostic evaluation and in whom lung transplantation (with removal of the native lung [lung explant]) was performed subsequently due to the development of end stage lung disease. Initial review of the clinical, physiologic, radiologic, and pathologic findings indicated a diagnosis of IPF in each patient based on the recent consensus statement for IPF.Citation3
Patient demographics, clinical history and physical exam findings, pulmonary function tests, and computed tomography images of the chest were reviewed in detail and findings documented for each patient. The time interval from SLB to lung transplantation was recorded for each patient.
Histologic evaluation
Following the initial chart review, a detailed qualitative and quantitative histologic evaluation was performed in each SLB and explant sample by a clinical pathologist (APB) with extensive experience in the histology of diffuse parenchymal lung disease. The presence and severity of the following findings were assessed as follows: geographic heterogeneity, lymphocytic aggregates, foci of smooth muscle bundles, fibroblastic foci, organizing pneumonia, pigmented macrophages, hyaline membranes, parenchymal fibrosis, and cystic spaces (honeycombing). A definitive pathologic diagnosis was rendered on each tissue sample according to standard criteria.Citation1–Citation3
Quantitative assessments were performed on the upper and lower lobe of each SLB and lung explant tissue sample for the following characteristics: (1) histologic fibrosis and honeycomb change, which were scored based on percentage involvement of the tissue sample (none, 1%–10%, 11%–25%, 26%–50%, >50%), and (2) fibroblastic foci and lymphocyte aggregates, which were scored as absolute numerical counts observed in total lung tissue and then standardized to 1 cm2 of lung tissue. Groups of lymphocytes were counted as an aggregate if ≥100 μm in diameter. To assist with quantitative scoring of samples, all sections were scanned in their entirety into a digital pathology system (Aperio Technologies, Vista, CA, USA), which allowed for analyzing slides at any magnification up to the highest magnification scanned.
Analyses of quantitative scores were performed by comparing paired, matched samples of SLB to lung explant samples. Specifically, each score for upper lobe SLB was compared to upper lobe explant, and each score for lower lobe SLB compared to lower lobe explant, for each individual patient. Our comparison analysis was ultimately limited by the number of SLBs available, due to the inherent risks of obtaining numerous SLB samples from IPF patients. All SLB scores (upper and lower lobe) were compared to all explant samples (upper and lower lobe) in a paired, matched format to provide the best representation of the histologic features in the entire lung.
Immunohistochemical evaluation
Immunohistochemistry was performed to further characterize the lymphocytic aggregates. For CD3, CD20, CD68, and Ki-67, pre-diluted monoclonal rabbit (CD3, Ki-67) and mouse (CD20, CD68) antibodies (Ventana, Tucson, AZ, USA) were utilized with an automated immunostainer and the Ultraview indirect biotin-free diaminobenzidine detection kit (BenchMark, Ventana, Tucson, AZ, USA). Polyclonal rabbit antibody against cleaved caspase 3 (EMD Millipore, Billerica, MA, USA) was used to histologically assess cellular apoptosis.
Following immunohistochemical staining for CD3 and CD20, morphometric analysis was performed to further assess the degree of CD3+ and CD20+ cell infiltration in tissue samples. Image thresholding and area measurements were performed using the ImageJ programCitation15 to determine total surface tissue area, percentage of tissue area occupied by CD3+ and CD20+ cells, and the ratio of tissue area occupied by CD3+ and CD20+ cells, respectively.
Statistical analyses
Differences between paired matched samples were analyzed by nonparametric Wilcoxon signed rank test (Graph Pad Software, La Jolla, CA, USA) or by paired two tailed Student’s t-test. Differences at P < 0.05 were considered significant.
Results
Patient characteristics
The patient cohort (n = 16) consisted of ten males and six females. There were 12 Caucasians and four African-Americans, and the median age [1st, 3rd quartiles] was 62 [57,66] years. Ten patients were lifelong nonsmokers, and no patient was an active smoker at the time of SLB or lung transplantation. For the entire cohort, pulmonary function testing indicated a moderately severe restrictive ventilatory defect, demonstrated by a forced vital capacity of 59 [44, 69] percent predicted, and a severe abnormality in gas transfer, demonstrated by a diffusing capacity for carbon monoxide of 45 [34, 55] percent predicted. Median time from SLB to subsequent lung transplantation in the cohort was 24 [15, 29] months.
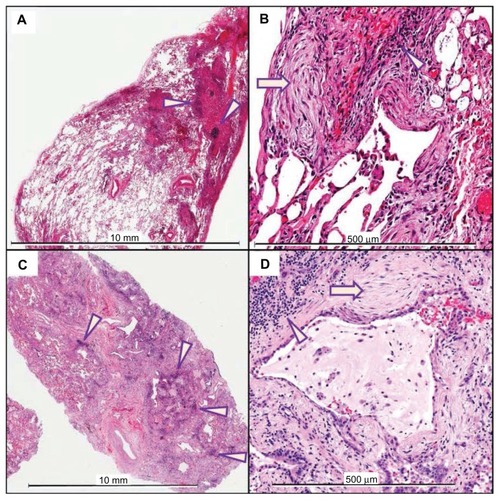
Detailed histologic analysis confirmed the pathologic pattern of UIP on both SLB and lung explant tissue in each patient. shows low and high magnification sections of the SLB (upper panels) and lung explant (lower panels) from a typical patient. The presence of UIP in conjunction with the clinical, physiologic, and radiographic findings confirmed the diagnosis of IPF in all patients.
Figure 1 Hematoxylin and eosin staining of SLB (A and B; scales indicated in each panel) and lung explant (C and D) from a typical patient with a 25 month interval between procedures.
Notes: Both the SLB and explant demonstrate a pathologic pattern of UIP, characterized by a patchwork pattern of fibrosis, architectural distortion, honeycomb change, fibroblastic foci (arrows), and aggregates of lymphocytes (arrowheads).
Abbreviations: SLB, surgical lung biopsy; UIP, usual interstitial pneumonitis.
Histologic analysis of fibrosis and honeycomb change
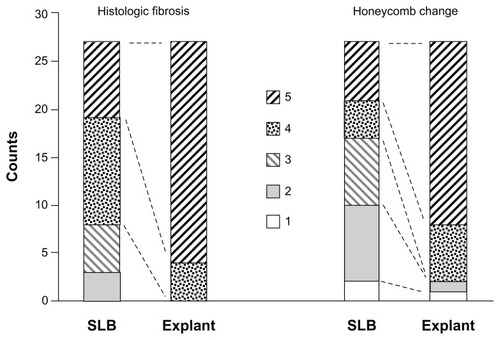
The percentage involvement of histologic fibrosis and honeycomb change was assessed in each SLB and explant tissue sample. Analyses of scores were performed by comparing paired, matched samples of SLB to lung explant samples. Specifically, scores for upper lobe SLB were compared to upper lobe explant, and scores for lower lobe SLB compared to lower lobe explant, for each individual patient. As shown in , there was more histologic fibrosis and honeycomb change in the explant samples compared with SLB across the cohort (P < 0.001 and P < 0.01, respectively, Wilcoxon signed rank test). These findings were consistent with the known progression of UIP in patients with IPF.
Figure 2 Stacked column graph showing the degree of histologic fibrosis and honeycomb change in SLB and explant samples, scored by percentage involvement of tissue: 1 (none), 2 (1%–10%), 3 (11%–25%), 4 (26%–50%), and 5 (>50%).
Notes: There was more histologic fibrosis and honeycomb change in the explant samples compared to SLB across the cohort (P < 0.001 and P < 0.01 respectively, Wilcoxon signed rank test).
Abbreviation: SLB, surgical lung biopsy.
Histologic analysis of fibroblastic foci and lymphocyte aggregates
The absolute number of fibroblastic foci and lymphocyte aggregates was counted in each SLB and explant sample, and then standardized to 1 cm2 of lung tissue. Analyses of counts were performed by comparing paired, matched samples of SLB to lung explant samples for each individual patient (upper lobe SLB to upper lobe explant and lower lobe SLB to lower lobe explant). For fibroblastic foci, there were equal numbers found in SLB and explant samples across the cohort (P = 0.74, Wilcoxon signed rank test).
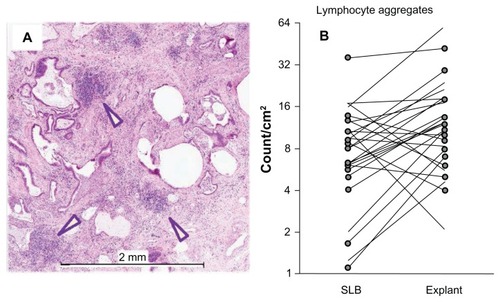
For lymphocyte aggregates, as shown in , there were more lymphocyte aggregates found in the explant samples compared to SLB. shows histology from a typical patient demonstrating selected aggregates of mononuclear cells ≥ 100 μm in diameter which were counted as lymphocyte aggregates. shows changes in the numbers of aggregates in the SLB and explant samples, and most notably, higher numbers of lymphocyte aggregates were observed in the explant samples compared to SLB across the cohort (P = 0.013, Wilcoxon signed rank test). This finding disputed the frequent assumption that inflammation decreases as disease progression occurs in patients with IPF.
Figure 3 Lung explant histology (A) from a typical patient demonstrating selected aggregates of mononuclear cells ≥ 100 μm in diameter (arrowheads) which were counted as lymphocyte aggregates. A scatter plot (B) composed of numerical counts of lymphocyte aggregates compares upper lobe SLB to upper lobe explant (unmarked ends) and lower lobe SLB to lower lobe explant (circled ends) for each individual patient.
Note: There were more lymphocyte aggregates in the explant samples compared to SLB across the cohort (P = 0.013, Wilcoxon signed rank test).
Abbreviation: SLB, surgical lung biopsy.
Immunohistochemistry was used to further characterize the composition of the lymphocyte aggregates, as shown in from a lung explant sample. The mononuclear cell aggregates consisted of CD3+ (T lymphocyte) and CD20+ (B lymphocyte) cells. Staining for CD68 showed very few CD68+ monocytes or macrophages within the lymphocyte aggregates, but did demonstrate CD68+ cells within small airways, alveolar spaces, and honeycombed cysts.
Figure 4 Immunohistochemistry of lung explant tissue for CD3 (A, B), CD20 (C, D), Ki-67 (E), and CD68 (F). The framed areas in the low magnification images for CD3 and CD20 are shown at higher magnification in the right panels (B and D, respectively). Ki-67, a marker of cellular proliferation, shows minimal positivity within the lymphocyte aggregates. The aggregates consist almost exclusively of T cells and B cells, with only a few CD68+ cells within the aggregates.
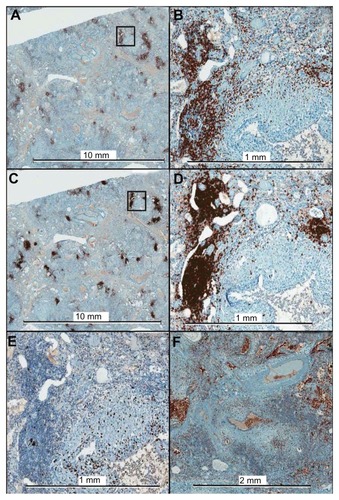
As can also be seen in , CD20+ cells were predominantly present in cohesive, focal clusters, whereas CD3+ cells were present not only in cohesive clusters, but also diffusely throughout the tissue as single cells or small groups of a few cells. Morphometric analysis was performed to further assess the degree of CD3+ and CD20+ cell infiltration in five explant tissue samples. There was a greater mean percentage of tissue area occupied by CD3+ cells (14.0 ± 7.4) than by CD20+ cells (7.2 ± 3.9) (P = 0.016, paired two tailed Student’s t-test). There was also a remarkably consistent ratio of the percentage area occupied by CD3+ and CD20+ cells, respectively, with a CD3+/CD20+ ratio of 2.0 ± 0.4 amongst the samples.
Staining for Ki-67, a nuclear antigen expressed during all active phases of the cell cycle and a marker of cellular proliferation, showed only minimal positivity within the lymphocyte aggregates. Staining for cleaved caspase 3, a marker for cellular apoptosis, showed no positive staining within the lymphocyte aggregates, but did show a few positive staining mesenchymal cells in the lung parenchyma, and a few positive staining granulocytes in small pulmonary vessels (data not shown). These results indicate the presence of nonproliferating, nonapoptotic lymphocytes within the aggregates.
Discussion
Our results indicate that as disease progression occurred in patients with IPF, histologic fibrosis and honeycomb change worsened as was expected,Citation6 but additionally, and contrary to current opinion,Citation5,Citation12–Citation14 aggregates of lymphocytes progressively accumulated in the lungs as well. To our knowledge, this is the first study which has objectively evaluated histologically the degree of lymphocytic infiltration in IPF over the duration of patient disease.
These findings are particularly important due to their divergence from the current dogma which states that inflammation decreases, and may even disappear (become “burned out”), as disease progression occurs in IPF.Citation5,Citation12–Citation14 Furthermore, a prevailing hypothesis has been put forward by many stating that inflammation has no role in the mechanisms or pathogenesis of IPF.Citation16–Citation18 The basis of these opinions most likely results from (1) a shift in IPF focus away from inflammation and towards aberrant wound healing, and (2) the ineffectiveness of traditional anti-inflammatory therapies, which will be discussed further below. The novelty of our study is the objective evaluation of inflammatory cell infiltration over the duration of patient disease. We found that across our IPF cohort, lymphocyte aggregates were present in greater numbers in advanced disease (explant tissue) compared to early disease (SLB), supporting the concept that active cellular inflammation continues in IPF even in severe end stage disease.
Our immunohistochemical results confirmed that lymphocyte aggregates consisted of CD3+ (T lymphocytes) and CD20+ (B lymphocytes) cells, with CD20+ cells predominantly located centrally within the aggregates, and CD3+ cells located more peripherally within aggregates and also diffusely throughout the lung tissue as single cells or groups of a few cells. In agreement with our findings, these lymphocyte aggregates have been previously described in patients with IPF as “ectopic organized lymphoid structures,” and have been shown to consist of nonproliferating, but activated CD3+ and CD20+ cells.Citation7 Mechanistic research from numerous laboratories has indicated that pulmonary lymphocytes may promote or alternatively attenuate fibrosis depending on their molecular phenotype.Citation11,Citation19–Citation23 Our morphometric analysis showed that despite the clustering of CD20+ cells within the aggregates, more total tissue area was occupied by CD3+ cells than by CD20+ cells, with a remarkably consistent ratio of approximately 2:1 amongst the samples. Increased numbers of CD3+ cells in patients with IPF has been described previously, and an inverse correlation has been demonstrated in IPF patients between the number of CD3+ cells and survival.Citation24,Citation25
Although lymphocyte aggregates were extensive in explant tissue, immunostaining for Ki-67 (a marker of active cell cycle cell proliferation) and for cleaved caspase 3 (a marker for cellular apoptosis) indicated that these lymphocytes were nonproliferating and nonapoptotic. The presence of abundant nonproliferating, nonapoptotic lymphocytes in these patients suggests that homing to the lung from the systemic circulation is the mechanism of lymphocyte accumulation, which is likely driven by a spectrum of cytokines and chemokines implicated in this process.Citation8,Citation9,Citation11
The precise etiology of IPF remains unknown despite four decades of intense research. Although numerous mechanisms have been explored, the most consistently studied mechanism has been the role of inflammation. Despite the universal presence of numerous pro-inflammatory cells, cytokines, chemokines, and cell surface molecules in these patients, the importance of inflammation has been recently challenged. One of the major arguments often cited for a noncausal role for inflammation is the ineffectiveness of anti–inflammatory therapies, particularly corticosteroids, in improving survival in patients with IPF.Citation3 However, it is important to note that resistance to corticosteroids does not always equate to the lack of important inflammatory mechanisms. There are several clinical pulmonary and nonpulmonary diseases in which inflammation is well established as the fundamental mechanism, but in which conventional anti–inflammatory therapy is poorly or completely ineffective.Citation26 One such example for this discrepancy is oxidative stress promoting corticosteroid resistant inflammation in the lung,Citation27 a particularly plausible concept in pulmonary diseases given the uniquely oxygenated environment of the lung, and the numerous observations implicating oxidative stress in mechanisms of pulmonary fibrosis.Citation11 Continued efforts to understand the precise role of inflammation and possible mechanisms of corticosteroid resistant inflammation remain warranted in IPF patients.
Despite some limitations, such as a relatively small sample size and observational nature of our measurements, the strength of our work is a unique set of matching lung tissue samples consisting of a surgical lung biopsy obtained early in the course of disease and lung explant tissue obtained at the time of end-stage disease. The ability to obtain matched samples such as these in patients with IPF is relatively infrequent due to appropriate caution in proceeding with surgical lung biopsy in IPF patients, and the inherent and frequent obstacles to successful lung transplantation. Given the well known heterogeneity of the lung in IPF, we compared all paired, matched samples (upper lobe SLB to upper lobe explant and lower lobe SLB to lower lobe explant) in the statistical analysis to achieve the most scientifically valid comparison, and to provide the best representation of the histologic features in the entire lung. The numerical assessment of histological changes in these matched samples allowed for our novel observation that greater numbers of lymphocyte aggregates were present in advanced disease compared with early disease in patients with IPF. Further mechanistic studies are warranted to determine the precise role of these infiltrating lymphocytes.
Disclosure
The authors report no conflicts of interest in this work.
Acknowledgments
Supported by Department of Veterans Affairs Merit Award 5I01CX000107 and National Institutes of Health – National Heart, Lung, and Blood Institute Grant 1R21HL106196 (both to SPA).
References
- American Thoracic Society, European Respiratory SocietyAmerican Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, Jun 2001 and by the ERS Executive Committee, Jun 2001Am J Respir Crit Care Med2002165227730411790668
- MyersJLKatzensteinALBeyond a consensus classification for idiopathic interstitial pneumonias: progress and controversiesHistopathology20095419010319054158
- RaghuGCollardHREganJJAn official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and managementAm J Respir Crit Care Med2011183678882421471066
- ScaddingJGHinsonKFDiffuse fibrosing alveolitis (diffuse interstitial fibrosis of the lungs). Correlation of histology at biopsy with prognosisThorax19672242913046035793
- CrystalRGFulmerJDRobertsWCMossMLLineBRReynoldsHYIdiopathic pulmonary fibrosis. Clinical, histologic, radiographic, physiologic, scintigraphic, cytologic, and biochemical aspectsAnn Intern Med1976856769788793466
- KatzensteinALZismanDLitzkyLNguyenBKotloffRUsual interstitial pneumonia: histologic study of biopsy and explant specimensAm J Surg Pathol200226121567157712459623
- Marchal-SomméJUzunhanYMarchand-AdamSCutting edge: nonproliferating mature immune cells form a novel type of organized lymphoid structure in idiopathic pulmonary fibrosisJ Immunol2006176105735573916670278
- HomerRJEliasJALeeCGHerzogEModern concepts on the role of inflammation in pulmonary fibrosisArch Pathol Lab Med2011135678078821631273
- WynnTAIntegrating mechanisms of pulmonary fibrosisJ Exp Med201120871339135021727191
- CrooksMGAslamIHartSPInflammation and pulmonary fibrosisKhatamiMInflammatory Diseases-Immunopathology, Clinical and Pharmacological BasesRijekaIntech201299128
- ToddNWLuzinaIGAtamasSPMolecular and cellular mechanisms of pulmonary fibrosisFibrogenesis Tissue Repair2012511122824096
- MeliconiRLalliEBorzìRMIdiopathic pulmonary fibrosis: can cell mediated immunity markers predict clinical outcome?Thorax19904575365402118691
- FlahertyKRTravisWDColbyTVHistopathologic variability in usual and nonspecific interstitial pneumoniasAm J Respir Crit Care Med200116491722172711719316
- StrieterRMPathogenesis and natural history of usual interstitial pneumonia: the whole story or the last chapter of a long novelChest20051285 Suppl 1S526S532
- RasbandWSImage JUS National Institutes of HealthBethesda, Maryland, USA1997–2012 http://imagej.nih.gov/ij/Accessed January 25, 2013
- SelmanMKingTEPardoAAmerican Thoracic Society; European Respiratory Society; American College of Chest PhysiciansIdiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapyAnn Intern Med2001134213615111177318
- GauldieJPro: Inflammatory mechanisms are a minor component of the pathogenesis of idiopathic pulmonary fibrosisAm J Respir Crit Care Med200216591205120611991866
- NoblePWHomerRJIdiopathic pulmonary fibrosis: new insights into pathogenesisClin Chest Med200425474975815564020
- LuzinaIGToddNWIaconoATAtamasSPRoles of T lymphocytes in pulmonary fibrosisJ Leukoc Biol200883223724417962367
- LuzinaIGToddNWNacuNRegulation of pulmonary inflammation and fibrosis through expression of integrins alphaVbeta3 and alphaVbeta5 on pulmonary T lymphocytesArthritis Rheum20096051530153919404954
- PochetuhenKLuzinaIGLockatellVChoiJToddNWAtamasSPComplex regulation of pulmonary inflammation and fibrosis by CCL18Am J Pathol2007171242843717569779
- LuzinaIGPapadimitriouJCAndersonRPochetuhenKAtamasSPInduction of prolonged infiltration of T lymphocytes and transient T lymphocyte-dependent collagen deposition in mouse lungs following adenoviral gene transfer of CCL18Arthritis Rheum20065482643265516868995
- AtamasSPYurovskyVVWiseRProduction of type 2 cytokines by CD8+ lung cells is associated with greater decline in pulmonary function in patients with systemic sclerosisArthritis Rheum19994261168117810366109
- DaniilZKitsantaPKapotsisGCD8+ T lymphocytes in lung tissue from patients with idiopathic pulmonary fibrosisRespir Res200568116042790
- ParraERKairallaRARibeiro de CarvalhoCREherECapelozziVLInflammatory cell phenotyping of the pulmonary interstitium in idiopathic interstitial pneumoniaRespiration200774215916917108669
- BarnesPJAdcockIMGlucocorticoid resistance in inflammatory diseasesLancet200937396781905191719482216
- ItoKHanazawaTTomitaKBarnesPJAdcockIMOxidative stress reduces histone deacetylase 2 activity and enhances IL-8 gene expression: role of tyrosine nitrationBiochem Biophys Res Commun2004315124024515013452