
Abstract
Background
Biliary atresia (BA) is a severe neonatal progressive cholangiopathy of unknown etiology. A timely Kasai portoenterostomy (KPE) improves survival of the native liver in patients with BA, although liver transplantation remains the ultimate treatment for most (60%–80%) patients. However, postoperative adverse effects of liver transplantation may be significant. In addition, patients require lifelong immunosuppressive therapy after liver transplantation.
Case Summary
Here, we report a case of a newborn female baby (birthday: 10–03-2018) with congenital BA (confirmed at 76 days of life) who survived KPE (first surgery at 85 days of life) and underwent successful living-related liver transplantation (LRLT) (second surgery at 194 days of life). Additionally, we reviewed the existing literature on BA. After KPE (at 85 days of life), the liver function of the baby did not improve, and the indicators of liver and kidney function showed a trend of aggravation, indicating that the liver function had been seriously damaged before KPE (at 85 days of life), demonstrating the urgent need for liver transplantation surgery. The female baby survived after part of her father’s liver was successfully transplanted into her body (at 194 days of life). The patient recovered successfully. No other diseases were found at the 4-year follow-up, and all indices of liver and kidney functions tended to be normal.
Conclusion
This case highlights the following. Postoperative alkaline phosphatase was consistently above the normal range, although the reason for this was unclear; neither tacrolimus nor cyclosporine A has formulations designed specifically for infants, which does not meet the needs of clinical individualized medication, suggesting that these anti-rejection drugs are future development directions. Only one case of congenital BA has been found thus far in Hefei, and this case has extremely important reference significance for the prevention, treatment, and diagnosis of BA in Hefei, Anhui province.
Graphical Abstract
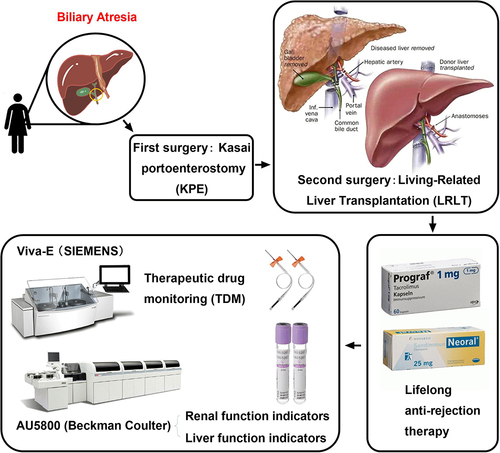
Introduction
Biliary atresia (BA) is a rapidly progressive biliary disease in children caused by fibrosis and obstruction of extrahepatic bile ducts, with a median survival time of 19 months.Citation1,Citation2 The disease manifests as neonatal jaundice and rapidly progresses to cholestatic liver fibrosis and cirrhosis within the first few weeks of life, making it the most common indication for pediatric liver transplantation worldwide; however, the pathophysiological basis of BA is currently unknown.Citation1–3 BA is mainly characterized by progressive fibrosis and inflammation of intrahepatic and extrahepatic bile ducts, resulting in bile duct blockage and cholestasis, which eventually leads to cholestatic cirrhosis and liver failure.Citation4 Newborn babies with BA often show yellow skin and sclera, pruritus, dysplasia, pale stool, dark urine, hepatosplenomegaly, and abdominal distension.Citation5 Successful treatment of BA requires the removal of the biliary obstruction as soon as possible. Of newborn babies with BA accompanied by bilirubin under 6 mg/dL who undergo Kasai portoenterostomy (KPE) under 30 days of age, 70% survive to two years of age with a native liver. However, this number drops to 50% and 0%, respectively, by age 90 days and >120 days.Citation6 Although the pathogenesis of BA is unknown, it may be related to autoimmune factors, virus infection, and toxin-mediated inflammation.Citation7 Investigators have recently revealed cases of BA in the same family and a higher incidence in some parts of the world (eg, Asian countries), suggesting that genetic predisposition contributes to the disease, and thus propose a genetic inheritance.Citation8 Modern genetic analysis has identified genetic variants responsible for BA, thus shifting the paradigm for explaining the phenotype of BA from acquired causes (eg, viruses, toxins) to genetically altered cholangiocyte development and function.Citation9 A recent genome-wide association study of 336 infants with non-syndromic BA and 8900 controls in China identified an association between an rs17095355 site in the ADD3 gene and the risk of BA (odds ratio (OR) = 1.70, 95% confidence interval (95% CI) = 1.49–1.99), although the signal of association between HLA genes and genetic susceptibility to BA is weak and may play only a small role in the genetic susceptibility to BA.Citation10 With the continuous progress shown in medical technology, the classification of BA has become increasingly accurate.Citation4,Citation7,Citation11 The current classification and frequency of BA phenotypes are as follows: isolated BA (67%–89%, accounting for the highest incidence by far), syndromic BA (4.9%–10%), cyst BA (5%–22.4%), and cytomegalovirus-associated BA (5%–9.5%).Citation7 The principal feature of isolated BA is biliary obstruction, followed by jaundice and cholestasis in the later stages.Citation7 Both KPE and liver transplantation are effective and successful treatments for BA, but they continue to present many challenges.Citation12 If surgery is not performed in time, most patients die within 2 years.Citation11 For infants with congenital BA, the age of operation is critical to the postoperative survival time and prognosis, as severe cholestasis can cause serious damage to the liver of a newborn baby.Citation6 Most studies have shown that delayed diagnosis and treatment of more than two months is generally associated with poorer natural liver survival for infants with congenital BA.Citation6 Recently, with the development of living-related liver transplantation (LRLT) technology and the improvement of human medical knowledge, living-related liver has become the main source of donor liver for babies with congenital BA.Citation13
Case Report
Even though there are a large number of extant studies on the prevention, treatment, and diagnosis of congenital BA in PubMed and ScienceDirect, we considered this topic novel, primarily because of the rarity of this disease.Citation4,Citation11,Citation14–16 The disease occurs on all continents, with variable geographic frequencies ranging from 1 in 5000 in Taiwan to an estimated 1 in 15,000 in the United States; no exact statistical data exist for the Chinese Mainland, and its incidence rate may be higher than 1/5000.Citation4,Citation11 BA is most common in East Asian countries, more common in Asia and Africa than in Europe, and more common in women than in men. French Polynesia has the highest rates (1:3500), with the chief reason related to intra-group marriage in isolated island populations; however, few epidemiological studies of BA have provided clues as to its etiology ().Citation13,Citation16 Our patient was a 4-year-old girl born on October 3, 2018, weighing approximately 3.8 kg. Neonatal jaundice was found at birth. Her mother underwent rigorous physical examination during pregnancy, and 4-dimension color Doppler ultrasonography of the fetal gallbladder at 24 weeks of gestation did not reveal any abnormal cysts. The baby had persistent jaundice after birth, liver function test results showed high levels of direct bilirubin (DBIL), indirect bilirubin (IBIL), and total bilirubin (TBIL) three weeks after birth, and was suspected of congenital BA by B ultrasound examination of the biliary tract 2.53 months after birth, which was confirmed by cholangiography at 76 days of life (According to Chinese expert consensus on BA, cholangiography is the most reliable diagnostic method for BA so far). With the extensive progress in medical and life sciences, the classification of BA has become increasingly accurate.Citation11,Citation15,Citation17 After careful re-review of our patient’s medical record, we ascertained that the patient’s disease was diagnosed as typical congenital BA, and that it reflected solid porta hepatis, because the most proximal part of the extrahepatic biliary tract within the porta hepatis of the patient was entirely solidified (). According to the classification criteria proposed by Hartley, the present case belongs to type 3, accounting for 90% of the total number of BA patients, and the case is also designated as typical type 3 (solid porta hepatis) according to the classification criteria proposed by Tomita ().Citation11,Citation17 The patient had no consanguineous marriage in her immediate three generations, no similar disease in her family history, and her immediate family members were all in good health, including her elder sister. Strikingly, the results of genetic testing showed no abnormalities, which is in agreement with previous studies, suggesting that this genetic detection technology cannot exclude congenital BA.Citation18 The clinical features, course, and outcome of BA have been well described. In newborn babies with BA, jaundice typically persists and the appearance of signs of cholestasis, such as dark urine and pale stools, follow.Citation18 The current diagnosis of congenital BA mainly includes clinical diagnosis, laboratory diagnosis, imaging diagnosis, and pathological diagnosis. The clinical diagnosis of congenital BA primarily includes white clay-colored stools, yellow brown urine, and jaundice; accompanied by abdominal distension, dysphoria, irritability, feeding difficulties, skin itching, and other manifestations (). Laboratory diagnosis primarily comprises serum levels of TBIL, DBIL, IBIL, ALT, AST, ALP, and coagulation function (). In addition, imaging diagnosis encompasses ultrasonography, MRI, CT, cholangiography, and nuclear medicine biliary imaging. The modalities commonly used in clinical cholangiography include percutaneous transhepatic cholangiography, endoscopic retrograde cholangiography, laparoscopy-assisted cholangiography, intraoperative cholangiography (IOC), intraoperative angiography, and postoperative angiography. Nuclear medicine hepatobiliary imaging constitutes the most advanced non-invasive method at present. By injecting a radioactive tracer into a vein, tracking the radiotracer can show the location and degree of BA. For example, a previous study reported that technetium 99m–diisopropylphenylcarbamoylmethyl-iminodiacetic acid (DISIDA) scintigraphy exhibited 100% sensitivity, but that the specificity of the DISIDA scan was only 70.4%.Citation19 Imaging examination is one of the important means to diagnose BA, while the chief pathological diagnosis is liver biopsy, and while liver biopsy is the most accurate method to diagnose BA, the disadvantage is that it generates trauma to the body.Citation20 In summary, BA can be diagnosed more accurately through the combination of clinical diagnosis, laboratory diagnosis, imaging diagnosis, and pathological diagnosis (). However, in our case, no white clay stools were observed. Otherwise, doctors can initially diagnose a baby with congenital BA in advance. This is inconsistent with former literature, which suggests that this indicator is not a characteristic indicator of the disease ().Citation7,Citation13 Before the first KPE surgery (at 85 days of life), all liver function indices well exceeded the normal value, suggesting severe liver dysfunction due to cholestasis (). The baby underwent KPE at the age of 2.83 months or 85 days of life. Unfortunately, the conditions were not improved after KPE. Therefore, at 194 days of life, the baby underwent the second LRLT surgery (), followed by lifelong immunosuppressive therapy after liver transplantation ( and ). In , at 113 days of life, we can see that the test results of serum TBIL (D), DBIL (E), IBIL (F), TBA (G), ALT (H), AST (I), GGT (J), ALP (K), and LDH (M) far exceeded the normal range (red straight bar). Additionally, serum ADA (C) could not be determined, likely because the result exceeded the detection limit of the advanced automatic biochemical analyzer in our hospital (Beckman Coulter, AU5800, USA). In contrast, UREA (A) and CREA (B) were below the normal range (green straight bar). CK was the only index that remained normal (L). Almost all the liver and kidney function indices of serum deviated from the normal range, but jaundice eased and CK remained normal. The baby had a chronic lack of appetite and stopped gaining weight, suggesting that preoperative cholestasis due to BA caused permanent and irreversible liver damage. Therefore, after consultation, the doctors decided that the patient was in urgent need of a liver transplantation.
Figure 1 Epidemiology of biliary atresia (BA) worldwide (words in red for emphasis).
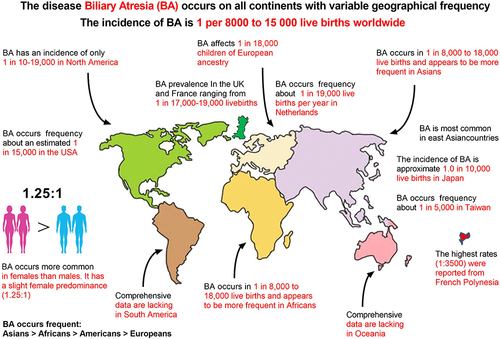
Figure 2 (A) Classification criteria of biliary atresia (BA). (B) Clinical presentation of BA (words in red for emphasis). (C) Diagnostic features in BA. (D) The present medical case belongs to type 3 (words in red for emphasis). (E) Developmental history of KPE and liver transplantation worldwide (words in red for emphasis).
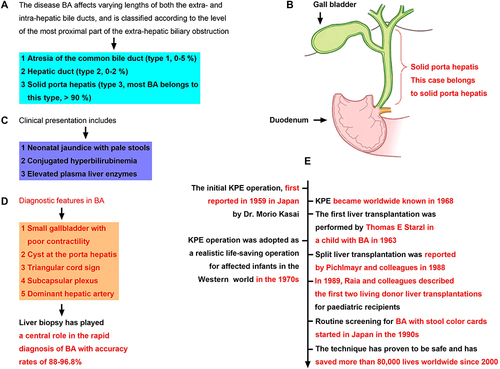
Figure 3 Clinical course of the baby and results of TDM for ISD. (A) Clinical history (timeline) of the baby (words in red for emphasis). (B) Dynamic whole-blood Tac valley concentration monitoring. ISDs are the key to anti-rejection treatment after organ transplantation in the effective therapeutic range. After liver transplantation, the ISD Tac is taken orally, but its therapeutic window is narrow and it is difficult to control its effective blood concentration with an oral dose. From 317 days of life to 383 days of life, the concentration of the drug in the blood of the baby far exceeded the normal range, resulting in renal toxicity (border colour in red for beyond the normal range). (C) Dynamic whole-blood CsA valley concentration monitoring shows a wide therapeutic window and an easily controlled oral dose of CsA. After dose adjustment, the effective blood concentration can be controlled within a stable range (black column) from 397 days of life to 1367 days of life, reducing adverse reactions to the liver and kidney (border colour in red for beyond the normal range, border colour in green for below the normal range).
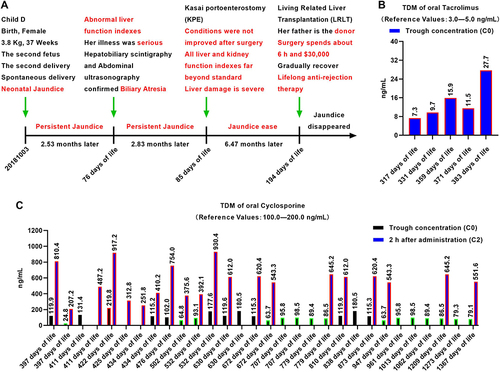
Figure 4 Postoperative dynamic monitoring of renal and liver function indices in serum. The renal function indices include UREA (A) CREA (B) and ADA (C) The liver function indices include TBIL (D) DBIL (E) IBIL (F) TBA (G), ALT (H) AST (I) GGT (J) ALP (K) CK (L) and LDH (M) Red indicates that the value is above the normal range, and green indicates that the value is below the normal range. Tac was changed to the oral ISD CsA at 397 days of life.
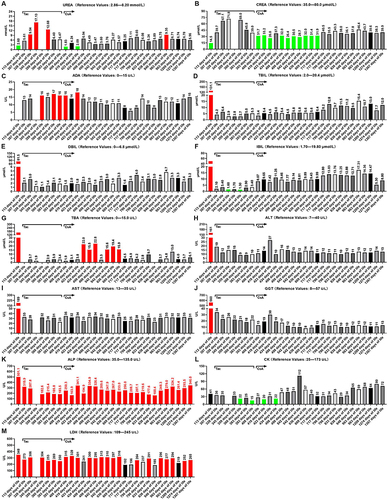
At 194 days of life, 6.47 months after birth, the baby underwent the second surgery, one of the biggest and most complicated surgeries of her life, LRLT, with her father as the donor. Doctors removed the patient’s badly damaged liver, then transplanted a portion of a healthy liver from her father into the baby’s body. Pathological findings revealed that the removed liver of the baby had been severely damaged by chronic cholestasis, with severe signs of liver fibrosis and cirrhosis (data not shown). The risky operation lasted 6 h and cost approximately $30,000. Fortunately, the transplantation was successful. After surgery, persistent jaundice resolved quickly and completely without oral ursodeoxycholic acid. Additionally, no glucocorticoids were used. In , on 307 days of life, the test results of serum UREA (A), CREA (B), ADA (C), TBIL (D), DBIL (E), IBIL (F), ALT (H), AST (I), GGT (J), and CK (L) were all within the normal range. Additionally, serum TBA (G) could not be determined, possibly because the newly transplanted liver was not yet fully functional in the patient’s body. In contrast, the levels of ALP (K) and LDH (M) were beyond the normal range, although the reason for this is unclear. An immunity inhibitor was administered to prevent acute rejection and its concentration in the blood was tested regularly ( and ). Almost all of the liver and kidney function indices of serum were in the normal range except for ALP () and LDH (). The patient gradually recovered and gained weight, suggesting that the second surgery (LRLT) was successful. After two major surgeries, the baby girl survived, and the clinical course is shown in .
The baby recovered gradually after surgery and was regularly given an immunity inhibitor to prevent acute rejection (Table S1). During administration, we found that Tacrolimus (Tac) had a narrow therapeutic window (3.0–5.0 ng/mL) (), and as a result, was replaced with cyclosporine A (CsA), with a wider therapeutic window (100.0–200.0 ng/mL). The dynamic blood concentrations of Tac and CsA in the baby are shown in and . Therapeutic drug monitoring (TDM) results showed that the baby’s blood concentration increased rapidly to the maximum value and exceeded the normal range 2 h after taking CsA orally, before decreasing slowly to the normal value (Table S1). However, as long-term high concentrations of CsA in the blood may cause adverse reactions, especially in the kidney, we dynamically recorded the renal function indicators (UREA, CREA, and ADA, Table S2).Citation21 The results showed that the serum concentrations of UREA, CREA, and ADA tended to be normal (). Previous studies have shown that long-term oral immunosuppressive CsA may cause diarrhea, although we noted no diarrhea induced by prolonged oral CsA. At present, there is no special oral sustained and controlled release preparation of CsA for babies, and the results suggest that this kind of preparation of anti-rejection drugs is a future development direction.
The liver function of the baby was severely impaired before the liver transplantation, possibly because a large amount of bile produced by the liver could not be excreted properly into the gallbladder, resulting in massive cholestasis in the liver, resulting in severe liver damage. Soon after the first operation at 113 days of life, the liver function indices including TBIL (), DBIL (), IBIL (), and TBA () were still far beyond the normal range, which may be due to the original liver damage or the fact that the donor liver was taken from an adult.Citation22 After the second operation (at 194 days of life), the liver function was tested regularly, and the results showed that these four indicators gradually tended to be normal, suggesting gradual recovery of both the liver function and the body. Additionally, three important indices of liver function, ALT (), AST (), and GGT (), gradually returned to normal after the second surgery-liver transplantation (at 197 days of life, Table S2). Dynamic monitoring results indicated that the newly transplanted liver gradually began to function normally in the baby, with normal bile secretion and excretion, and successful LRLT.
Dynamic monitoring showed that the baby’s postoperative ALP was consistently above the normal range, but tended to decline overall (, Table S2). Although the reason for this remains unclear, it may be due to incomplete recovery of transplanted liver function.Citation23 Additionally, previous studies have found that ALP often exceeds the normal range during infant development, and the higher ALP in this case may also be related to the rapid growth of the body.Citation24 Moreover, four dynamic monitoring test results showed that CK in blood was below normal (green straight bar), but the long-term results tended to be normal (). Furthermore, another biochemical index of liver function, LDH, in the baby’s blood was beyond the normal range, but generally tended to decline, and may be restored to the normal range in the future (). In conclusion, the baby recovered well after liver transplantation.
Interestingly, several postoperative blood routine tests revealed mild anemia in the infant, which has not been reported in previous studies (at 1274 days of life). This is a rare adverse effect of oral CsA. We do not know the specific cause of the anemia, but it may be related to the long-term use of immunosuppressive drugs (ISDs). Additionally, previous studies have shown that CsA binds to blood cells and plasma proteins at a high rate, and CsA is rapidly distributed into erythrocytes after oral administration, with 60%–75% taken up by blood cells; however, thus far, there have been no reports of CsA destroying erythrocytes. Furthermore, hematocrit levels were lower than the normal range in some blood routine tests.
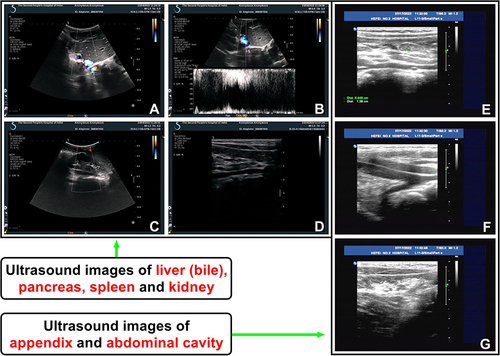
The results of abdominal color Doppler ultrasound at 1298 days of life showed that the transplanted liver was complete in shape and smooth in the surface, with a portal vein flow rate of 51 cm/s, suggesting that the transplanted liver played a normal role in the body (). The follow-up was conducted at 1383 days of life, during which abdominal color Doppler ultrasound revealed no lump, seroperitoneum, and abnormal flow in the abdominal cavity and/or around the appendix (). Therefore, no lump and other abnormalities caused by long-term use of ISD were found in the major organs and/or tissues of the abdomen.
Figure 5 Abdominal color Doppler ultrasonography showed normal transplanted liver, bile, and renal function (at 1298 days of life), and no lump, seroperitoneum, and abnormal flow in the abdominal cavity and/or around the appendix (at 1383 days of life) (words in red for emphasis). (A and B) The transplanted liver was regular in shape, smooth in surface, homogeneous in parenchymal echo and even in light spot. The portal vein blood flow velocity at the portal was 51 cm/s. The inner diameter of the inferior vena cava was not wide and the inner diameter of the common bile duct was not expanded. (C and D) Both kidneys were normal in shape and size, with a clear outline, complete and smooth renal capsule, uniform echo in parenchyma, and concentrated echo in the collecting system. No obvious abnormal echo was observed. (E–G) Abdominal color Doppler ultrasound revealed no lump, seroperitoneum, and abnormal flow in the abdominal cavity and/or around the appendix.
In conclusion, all liver function indices of the baby’s blood after KPE and LRLT tended to be normal except for ALP, and the test results indicated that the LRLT was successful. Patients undergoing liver transplantation should be treated with lifelong ISD. Currently, the most commonly used ISDs are Tac and CsA. Here, CsA was regularly taken orally and the blood concentration was tested in parallel to maintain a normal blood concentration level and reduce adverse reactions (Table S1). After adjusting the ISD dosage, the effective blood concentration was maintained within the normal range, and the adverse reactions caused by long-term drug use were reduced. No abnormalities were found in the renal function monitoring indicators. This present case is significant since there are eight million residents in Hefei, but only one case of congenital BA has been discerned thus far. At present, the patient is recovering well after two major operations and is now approximately six years of age. Therefore, the occurrence of this case reflects extremely important reference significance for the prevention, treatment, and diagnosis of congenital BA in Hefei, Anhui province.
Discussion
Diagnosis of BA
BA is typically seen in full-term, normal birth-weight infants, and is characterized by jaundice shortly after birth, with lighter stool color and darker urine color. Abdominal B ultrasound showed an enlarged liver without bile duct dilatation and no gallbladder or small gallbladder observed after 4 h of fasting. However, a normal gallbladder does not rule out BA. Ultrasonography of hilar fibrous mass (triangle sign) is a specific manifestation of BA, but depends on the experience of the operator, with a sensitivity of 49%–75%. Isotope hepatobiliary imaging, duodenal fluid drainage, liver biopsy, and pathologic endoscopic retrograde cholangiopancreatography are also helpful in differential diagnosis. If external BA cannot be ruled out after the above examination, laparoscopic or open cholangiopancreatography should be performed in time.
Patients Undergoing Liver Transplantation Must Take Lifelong ISD to Prevent Rejection
Congenital BA is characterized by the obstruction of bile ducts inside and outside the liver, which can lead to biliary cirrhosis in babies and eventually liver failure.Citation1,Citation4 At present, KPE is the preferred treatment for BA.Citation5,Citation6 However, the effect of KPE is directly related to the age of the baby at the time of the operation because the earlier the operation, the better the effect. Previous studies have demonstrated that KPE surgery should be performed before the first 3 months of life, preferably with a definitive diagnosis before 2 months of life, and KPE surgery performed within 45 days (1.5 months) of age can achieve the best bile flow recovery rate and long-term survival rate. Additionally, infants with BA who receive KPE alone may still require liver transplantation before adulthood because of previous severe liver damage. In this case, the baby was diagnosed with congenital BA 76 days (2.53 months) after birth and underwent KPE surgery 85 days (2.83 months) after birth. The liver and kidney function of the baby did not improve but worsened after the first surgery (KPE) because of the delay in the optimal operation time, indicating that the liver had been seriously damaged before KPE (Table S2). Therefore, the doctor decided to perform liver transplantation after consultation. It is necessary to maintain sufficiently high vigilance for BA to make a clear diagnosis and perform surgery as soon as possible. Liver transplantation is the most effective treatment for congenital BA in babies.Citation7,Citation11 It has been reported that the survival rate of babies after transplantation is close to 90%, and the earlier a liver transplantation is performed, the better the effect.Citation11,Citation13 However, complications after liver transplantation in babies still cannot be ignored or avoided.Citation21,Citation22 How to effectively prevent and treat complications after liver transplantation in babies remains a clinical concern.
ISD Against Acute Rejection Reaction
Acute rejection reaction is the most common rejection reaction and one of the most serious complications after liver transplantation. Acute rejection reaction, which is seen after liver transplantation, usually occurs any time between the 5th and 15th days after surgery.Citation25 If the post-liver transplantation patient has recurrent fever, increased bilirubin and aminotransferase, and persistent abdominal distention after surgery, acute liver rejection should not be excluded.Citation26 Tac, mycophenolate mofetil, and intravenous administration of methylprednisolone can be used for acute rejection reactions.Citation27 Here, as the above symptoms did not occur when the baby regularly took the ISD Tac after liver transplantation, acute rejection reaction was excluded. On the FDA website, the biomarker for Tac is CYP3A5*3. For routine clinical application of Tac, the steady-state serum C0 of Tac should be adjusted to the effective concentration range under the guidance of TDM. Currently, the information provided by the FDA indicates that determination of the CYP3A5 genotype before transplantation improves understanding of the metabolic characteristics of Tac before surgery, which allows the most appropriate drug and dose to be selected in the early postoperative initial treatment and ensures that the concentration of Tac can be adjusted to the effective concentration once or twice. The goal is to maximize the efficacy of the drug.
CsA is a potent ISD that can reversibly act on lymphocytes, inhibit cell-mediated immune responses (including alloimmune response), and inhibit the synthesis and release of lymphocytes, blocking stationary lymphocytes in the early G0 and G1 phases of the cell cycle. In this way, the survival time of allografts can be prolonged, and humoral and host immunity can be inhibited. CsA does not inhibit erythropoiesis and does not affect the function of phagocytic cells. Common side effects of CsA include anorexia, nausea, vomiting and other gastrointestinal reactions, gingival hyperplasia, and occasionally hyperglycemia anemia hyperuricemia. After kidney transplantation, in the short term, the erythropoietin in the body cannot reach a normal level, which affects the hematopoietic function of the bone marrow, resulting in a low number of red blood cells (RBCs) and low hemoglobin (HGB) content. Here, the recent blood routine examination revealed that the RBC, HGB, HCT, and mean platelet volume of the baby were all at lower-than-normal levels (at 1273 days of life), although the reason is unclear, and further study is needed.
The oral absorption of CsA is irregular and incomplete, and individual differences are large. The oral peak time of CsA is about 3.5 h, and it is mainly absorbed in the small intestine. The bioavailability of CsA is approximately 30% and can be increased with the extension of treatment time and the increase in drug dose. Absorption of CsA can be reduced in patients with liver disease or gastrointestinal dysfunction after liver transplantation. The binding rate of CsA with plasma protein is up to 90% (mainly binding with plasma lipoprotein). After absorption, CsA is distributed in all tissues of the body, mostly in the fat, liver, pancreas, lung, kidney, renal gland, spleen, and lymph nodes (the concentration in these tissues is higher than that in plasma). In the blood, 33%–47% of CsA is distributed in plasma, 4%–9% in lymphocytes, 5%–12% in granulocytes, and 41%–58% in RBCs. CsA is metabolized by the liver and affected by the P450 enzyme; it is almost completely metabolized in the body. CsA has at least 15 metabolites, some of which have immunosuppressive activity. CsA is mainly excreted in the intestine with bile (94%) and in the stool, while only 6% is excreted by the kidneys, and less than 0.1% of the original substance is excreted from the urine. The plasma half-life of CsA is 19 (10–27) h for adults and only 7 (7–19) h for children.
Selection of ISD in This Case
Calcineurin inhibitors (CNIs), represented by Tac and CsA, are the most widely used ISDs in clinical practice.Citation28 CNIs play their roles by inhibiting T cell activation and mixed lymphocyte reaction.Citation28 The inhibitory effect of Tac is more than 10 times that of CsA, but as the liver toxicity of Tac is lower than that of CsA, Tac is the first choice in clinical practice.Citation29 However, both Tac and CsA can cause hepatotoxicity and nephrotoxicity.Citation29 Here, the baby was given a normal dose of Tac regularly after surgery, but the results of TDM showed that the blood drug concentrations were all above the normal range (Table S1). Additionally, the therapeutic window of Tac is narrow (3.0–5.0 ng/mL) (Instruction of Siemens Healthcare Diagnostics Inc), which makes it difficult to control the dosage of infants.Citation30 Therefore, Tac was replaced with regular oral doses of CsA, which has a wide normal effective range of blood drug concentration (100–200 ng/mL) (Instruction of Siemens Healthcare Diagnostics Inc) and an easily controlled dosage in babies (Table S1).Citation31 To prevent liver and kidney toxicity caused by ISD, we recorded all serum indices of liver and kidney function in the baby in detail. Serum UREA and CREA increase when renal function is impaired (Table S2). Here, from 307 days of life to 393 days of life, serum UREA continued to increase after oral Tac (). Additionally, most test results of serum CREA were within the normal range after oral Tac, but all were at high levels from 307 days of life to 393 days of life (). On 359 days of life, both serum UREA and CREA could not be determined, likely due to the levels far exceeding the detection limit. Serum UREA and CREA are important indicators of renal function injury induced by drugs. These results suggest that oral administration of the ISD Tac during the postoperative period from 307 days of life to 393 days of life impaired the renal function of the baby. After changing to oral ISD CsA at 397 days of life, the serum UREA and CREA were mostly in the normal range and tended to be completely normal in the later stage from 408 days of life to 1367 days of life (Table S2). Many studies have reported that the nephrotoxicity of Tac is lower than that of CsA; however, we found the opposite result, in that these two renal function indices tended to be normal after long-term oral administration of CsA. We do not know the exact reason for this, which may be related to individual differences in medication. ADA is an enzyme that hydrolyzes adenosine in vivo and is central to the normal function of the immune system. Our previous studies found that inhibition of the adenosine pathway could alleviate alcoholic liver fibrosis in rats.Citation32 Here, the serum ADA levels tended to be normal when CsA was used instead of Tac after 397 days of life, suggesting that CsA was less toxic to the immune system ().
Interestingly, most of the seven tested serum liver function indices, including TBIL (), DBIL (), IBIL (), ALT (), AST (), GGT (), and CK (), were within the normal range (Table S2). However, when Tac was replaced with CsA, the serum liver function indices were more stable in the normal range, especially those of TBIL (), DBIL (), ALT (), AST (), and GGT (). Additionally, oral ISD Tac showed that four test results of serum IBIL () and CK () were below the normal range (green straight bar), and after switching to CsA, only two test results were below the normal range. Additionally, the test results of serum AST () were stable and within the normal range. These results suggest that the ISDs Tac and CsA have little effect on the liver function of the baby, and that the transplanted liver gradually plays a normal function in the body. Serum TBA is mainly used in the diagnosis of various liver diseases and is one of the most sensitive indicators of liver function. When hepatocytes are damaged, they cannot effectively absorb bile acids from the intestine, resulting in increased concentrations of TBA. Here, from 434 days of life to 669 days of life, we found that six test results of serum TBA () were above the normal level (red straight bar). Although we do not know the exact reason for this result, it may be related to the temporary increase in serum TBA level caused by the short-term high-protein diet of the baby. Therefore, the diet and nutrition of patients after liver transplantation is another important research direction. From 717 days of life to 1367 days of life, all test results of serum TBA were at the normal level (Table S2). It is clear that long-term oral CsA is less toxic to the liver, and that the transplanted liver gradually functions normally in the body of the baby.
BA Survival and Five-Year Survival
A successful KPE delays the need for an emergency liver transplant in early infancy and not only offers the potential for longer native liver survival (NLS) into adulthood but also provides valuable time in the search for a suitable liver donor.Citation13 Anouti et al conducted an updated assessment of 1052 transplanted pediatric and adult BA patients in the United States from 2010 to 2021 using the UNOS database, and their results showed that there were 441 (20.0%) living donor liver transplantations (LDLTs) and 611 (27.7%) split deceased-donor liver transplantations.Citation33 In the United States, the number of annual surgeries for BA liver transplantation was stable (an average of 182±16), while the number of surgeries for BA LDLT rose from 28 in 2010 to 55 in 2021, and the five-year graft survival rate for BA patients was 88.3% compared with 79.5% for non-BA elderly patients.Citation33 Five-year observation and relative survival estimates are widely used in studies of patients with congenital BA and liver transplantation as they provide a general description of disease outcomes and are useful for monitoring and comparative purposes.Citation15,Citation34,Citation35 Previous research by Chardot et al has revealed that BA patients exhibit a 30% chance of reaching adulthood without liver transplantation, and several large retrospective studies in Japan and other Western countries have focused on the long-term outcomes of liver transplantation for BA, reporting five-year and 10-year graft survival rates of 68.0%–98.0% and 71.0%–90.0%, respectively. An estimated 50% (20%–73%) of late-onset acute rejection and 15% (3%–35%) of transplant failures are associated with poor medication adherence after liver transplantation.Citation34 Chardot et al reviewed all 1107 BA patients born in France between 1986 and 2009 and found that 1044 (94%) BA patients had undergone KPE surgery and 38% of BA patients had complete abrogation of jaundice (serum TBIL≤ 20 μmol/L).Citation35 Results showed that five-year, 10-year, and 20-year NLS rates after KPE surgery were 40%, 36%, and 30%, respectively; and that the NLS rates at one, two, three months and 20 years after KPE surgery were 39%, 32%, 28%, and 19%, respectively. A total of 588 children received 692 liver transplantations, and the overall survival rates for BA patients at five, 10, and 20 years were 81%, 80%, and 77%, respectively.Citation35 As reported by Matsui, a program that screened 313,230 infants in Japan’s Tochigi Prefecture between 1994 and 2011 demonstrated NLS rates of BA children at five, 10, and 15 years of 88%, 77%, and 49%, respectively.Citation15 Authors of a Chinese population-based study estimated clinical data from 244 consecutive patients who underwent KPE and found that the overall median NLS rates were 41.2 months (95% CI, 30.8–51.6 months), and that the one-year, three-year, and five-year NLS rates were 85.4%, 61.1%, and 43.3%, respectively; with the conditional survival of patients with BA non-linear over time after KPE surgery.Citation36 These data necessitate large-scale, multicenter population studies to further follow up BA survivors and investigate their five-year survival with or without liver transplantation. At present, the long-term prognosis of BA patients with LDLT remains quite favorable.
Approximately 80% of Patients with BA Require Subsequent Liver Transplantation
Dong et al reported in 2018 on approximately 400 infant patients who were diagnosed with BA each year at the Children’s Hospital of Fudan University, one of the largest pediatric hospitals in China, and their study showed that the two-year survival rate of primary liver survival was 53.7% in BA patients, and the remaining 46.3% BA patients required subsequent liver transplantation.Citation37 An early investigation conducted by Mark et al surveyed all 148 infants with BA between January 1999 and June 2002 in England and Wales of the UK, and these authors found that a primary KPE was performed in 142 infants (with a median age at surgery of 54 [range 7–175] days) and a primary liver transplantation in five (with a median age at surgery of 248 [190–398] days); 135 babies were alive, 84 (62%) of whom had their native liver and 51 (38%) underwent transplantation.Citation38 Tessier et al also reported in 2019 that the outcomes after KPE were uneven, with 50% of infants needing a liver transplantation by 24 months, making BA the leading indication for pediatric liver transplantation.Citation39 Jain et al retrospective observational study identified a total of 397 patients with BA who underwent KPE procedure at King’s College Hospital between 1980 and 1996, and they found that patients with BA were at risk of needing a liver transplantation when beyond 16 years of age; their research further demonstrated 20-year NLS rates had been documented as 14–44% worldwide, but that most long-term native liver survivors developed complications in adulthood—including portal hypertension (PHT) and cholangitis, with reported liver transplantation rates as high as 22%. The higher end of the Jain study range came from Japan, where there was a longer experience of KPE and where transplantation practice is vastly different ().Citation40 Rebecca et al reported that approximately 80% of patients with BA will require liver transplantation, accounting for 50% of all pediatric liver transplantations.Citation41 Lee et al conducted a study at the National Taiwan University Children’s Hospital from 2015 to 2023 and enrolled 42 BA patients before and after KPE at approximately 2–9 months. These authors concluded that in Taiwan about 50% of patients with BA who undergo KPE eventually develop various advanced liver diseases and need liver transplantation before two years of age.Citation42 Authors of a previous study applied the Dutch national database for BA collected data of 104 BA patients on clinical features, liver biochemical indices, and liver ultrasonography from all BA patients who underwent KPE without liver transplantation and who survived to age 20 (n = 28; 27% of all 104 BA patients).Citation43 These researchers found that ≥25% of patients with BA survived at least 20 years without liver transplantation in the Netherlands, that female BA patients manifested a diminished perception of their health, and that 20% of long-term survivors remained asymptomatic, with no clinical or ultrasonographic signs of cirrhosis or portal hypertension.Citation43 A 2013 study by Chardot et al showed that BA patients had an 89% life expectancy and a 30% chance of surviving to adulthood without liver transplantation.Citation35 Early KPE, with no age threshold, reduced the need for liver transplantation before adulthood. BA accounts for about 50% of the indications for liver transplantation in children, and a French study of 1107 children with BA from 1986 to 2009 revealed that the majority of long-term survivors of BA patients showed native liver cirrhosis, and about 85–90% of BA patients who survived after KPE were likely to require liver transplantation in adulthood. Nevertheless, there is still a small percentage of BA patients (approximately 10–15%) who may never need liver transplantation.Citation35 Therefore, large multi-center investigations and studies are still needed in the future to further confirm the number of BA patients who require liver transplantation and to provide attention to patient survival status.
Monitoring Protocol of Trough Concentration (C0) and Two Hours After CsA Administration
At present, the monitoring scheme of CsA usually adopts the monitoring scheme of C0, as the TDM protocol of CsA has not yet reached consensus.Citation44–46 The monitoring protocol is an earlier and accepted protocol that refers to the lowest blood concentration after ingestion.Citation45,Citation46 Since CsA is routinely administered twice a day, the protocol refers to when the blood concentration of CsA reaches a steady state (usually on Day 2); ie, the whole blood concentration 12 hours after the first dose and before the second dose.Citation45,Citation46 In theory, the concentration at this time should be the lowest after each administration. Therefore, as long as the blood concentration of CsA is guaranteed within the treatment window, rejection reactions will not occur.Citation47 Based on this, the monitoring scheme is also the classic TDM regimen for CsA in clinical practice.Citation47,Citation48 The individual variation of CsA is large, and there are many influencing factors. To produce sufficient immunosuppressive effect in the narrow treatment window of transplant patients with the lowest adverse reactions, investigators of international prospective studies have proposed a new monitoring method called C2 (the concentration measured in whole blood about 2 hours after oral administration of the drug) monitoring method and hypothesize that adjusting the dosage of CsA based on C2 monitoring would significantly reduce the incidence of rejection, decrease the severity of acute rejection reaction, and generate a better predictive effect on acute renal toxicity of CsA than simply measuring C0.Citation49,Citation50 For liver transplantation patients, the effective concentration of CsA as recommended by C2 monitoring method is 1000 ng/mL at 0–3 months after surgery, 800 ng/mL at 3–6 months, and 600 ng/mL after 6 months.Citation46,Citation49 In summary, both basic research and clinical studies demonstrate that the C2 monitoring method of CsA is safe, effective, and economical in liver transplantation recipients. After consultation, both doctors and clinical pharmacists assigned the blood concentration monitoring protocol that measured both trough and 2-hr after CsA ingestion for the patient.
We uncovered no abnormality in the current case except for ALP, and the baby recovered well after liver transplantation surgery. Neither Tac nor CsA formulations are currently designed specifically for infants, and this does not meet the needs of clinical individualized medication. Therefore, it is common to use oral tablets and capsules in fractionated doses to compensate for inadequate dosage and specifications of existing medications. We recommend that precise adjustment of the clinical dose would be helpful in ensuring that the blood concentration of the recipient remains stable and within the target range.
Conclusions
At present, cholangiography remains the most accurate method to diagnose congenital BA, but this method can result in trauma to the body. Currently, KPE is the most effective method for treating congenital BA, but it is best to perform the surgery within 1.5 months after birth, and the earlier it is performed, the better the effect and the longer survival time. Many patients with congenital BA who underwent KPE in early childhood still require liver transplantation as adults, possibly due to permanent liver damage caused by cholestasis before surgery. Here, because the diagnosis was made 76 days after birth, the effect of KPE on the 85th day after birth was poor, and all postoperative liver and kidney indices were beyond the normal range, indicating that a liver transplantation was necessary. The patient recovered well after liver transplantation 6.47 months after birth. Regular oral ISDs were used to prevent rejection reaction and regular follow-up was conducted. In this case, it was found that an oral dose of 70 mg/day of CsA for children under 4 years of age can stabilize the blood concentration of CsA within the effective range and minimize the possible adverse reactions caused by long-term use. This case provides valuable experience for the prevention, diagnosis, and treatment of neonatal congenital BA.
Abbreviations
ADA, Adenosine Deaminase; ALP, Alkaline Phosphatase; ALT, Alanine Aminotransferase; AST, Aspartate Aminotransferase; BA, Biliary Atresia; CK, Creatine Kinase; CNI, Calcineurin inhibitor; CREA, Creatinine; CsA, Cyclosporine A; DBIL, Direct Bilirubin; GGT, Gamma-Glutamyl Transpeptidase; HCT, Hematocrit; HGB, Hemoglobin; IBIL, Indirect Bilirubin; ISD, Immunosuppressive drug; KPE, Kasai Portoenterostomy; LDH, Lactate Dehydrogenase; LRLT, living-related liver transplantation; RBC, Red Blood Cell; Tac, Tacrolimus; TBA, Total Bile Acid; TBIL, Total Bilirubin; TDM, Therapeutic drug monitoring.
Ethical Approval and Consent to Participate
The patient’s mother had given her consent for the article to be published. This article was performed in accordance with the principles of the Declaration of Helsinki, and the Hospital Ethics Committees of Hefei Hospital Affiliated to Anhui Medical University (The Second People’s Hospital of Hefei) approved the publication of case details.
Consent to Publish
The patient’s mother had given her consent for the article to be published.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that this research was conducted in the absence of any commercial or financial relationships that could be interpreted as a potential conflict of interest.
Acknowledgments
We thank Hefei Hospital Affiliated to Anhui Medical University (The Second People’s Hospital of Hefei) for providing the original data. We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript. We are immensely grateful to our generous patient’s mother for allowing us to publish their case.
Data Sharing Statement
The original contributions presented in the study are all included in the article/Supplementary Material, and further inquiries can be directed to the first author.
Additional information
Funding
References
- Bass LM, Ye W, Hawthorne K. et al. Risk of variceal hemorrhage and pretransplant mortality in children with biliary atresia. Hepatology. 2022;76(3):712–726. doi:10.1002/hep.32451
- Lim YZ, Zhu M, Wang Y, et al. Pkd1l1-deficiency drives biliary atresia through ciliary dysfunction in biliary epithelial cells. J Hepatol. 2024;24:00151–X. doi:10.1016/j.jhep.2024.02.031
- Trampert DC, Beuers U. A beneficial response of fetal wound healing gone bad in the bile duct: the overarching cause of biliary atresia. J Hepatol. 2024;80:387–389. doi:10.1016/j.jhep.2023.12.018
- Asai A, Miethke A, Bezerra JA. Pathogenesis of biliary atresia: defining biology to understand clinical phenotypes. Nat Rev Gastroenterol Hepatol. 2015;12(6):342–352. doi:10.1038/nrgastro.2015.74
- Liao FM, Chang KC, Wu JF, Chen HL, Ni YH, Chang MH. Direct bilirubin and risk of biliary atresia. Pediatrics. 2022;149(6):e2021053073. doi:10.1542/peds.2021-053073
- Jagadisan B, Dhawan A. Emergencies in paediatric hepatology. J Hepatol. 2022;76(5):1199–1214. doi:10.1016/j.jhep.2021.12.027
- He L, Chung PHY, Lui VCH, Tang CSM, Tam PKH. Current understanding in the clinical characteristics and molecular mechanisms in different subtypes of biliary atresia. Int J Mol Sci. 2022;23(9):4841. doi:10.3390/ijms23094841
- Sîrbe C, Benţa G, Pop TL. Immune-mediated cholangiopathies in children: the need to better understand the pathophysiology for finding the future possible treatment targets. Front Immunol. 2023;14:1206025. doi:10.3389/fimmu.2023.1206025
- Hellen DJ, Karpen SJ. Genetic Contributions to Biliary Atresia: a Developmental Cholangiopathy. Semin Liver Dis. 2023;43(03):323–335. doi:10.1055/a-2153-8927
- Cui MM, Gong YM, Pan WH, et al., Contribution of ADD3 and the HLA genes to biliary atresia risk in Chinese. Int J Mol Sci. 2023;24(19):14719. doi:10.3390/ijms241914719
- Hartley JL, Davenport M, Kelly DA, atresia B. Biliary atresia. Lancet. 2009;374(9702):1704–1713. doi:10.1016/S0140-6736(09)60946-6
- Lakshminarayanan B, Davenport M. Biliary atresia: a comprehensive review. J Autoimmun. 2016;73:1–9. doi:10.1016/j.jaut.2016.06.005
- Schreiber RA, Harpavat S, Hulscher JBF, Wildhaber BE. Biliary atresia in 2021: epidemiology, screening and public policy. J Clin Med. 2022;11(4):999. doi:10.3390/jcm11040999
- Glessner JT, Ningappa MB, Ngo KA, et al. Biliary atresia is associated with polygenic susceptibility in ciliogenesis and planar polarity effector genes. J Hepatol. 2023;79(6):1385–1395. doi:10.1016/j.jhep.2023.07.039
- Verkade HJ, Bezerra JA, Davenport M, et al. Biliary atresia and other cholestatic childhood diseases: advances and future challenges. J Hepatol. 2016;65(3):631–642. doi:10.1016/j.jhep.2016.04.032
- Kelly D, Samyn M, Schwarz KB, Biliary atresia in adolescence and adult life: medical, surgical and psychological aspects. J Clin Med. 2023;12(4):1594. doi:10.3390/jcm12041594
- Tomita H, Masugi Y, Hoshino K, et al. Long-term native liver fibrosis in biliary atresia: development of a novel scoring system using histology and standard liver tests. J Hepatol. 2014;60(6):1242–1248. doi:10.1016/j.jhep.2014.01.028
- Bezerra JA, Wells RG, Mack CL, et al. Biliary atresia: clinical and research challenges for the twenty-first century. Hepatology. 2018;68(3):1163–1173. doi:10.1002/hep.29905
- Lee SY, Kim GC, Choe BH, et al. Efficacy of US-guided percutaneous cholecystocholangiography for the early exclusion and type determination of biliary atresia. Radiology. 2011;261(3):916–922. doi:10.1148/radiol.11110665
- Mullany K, Arbuckle S, Mei-Ling Siew S. A rare cause of neonatal cholestasis without liver dysfunction. Gastroenterology. 2019;156(8):e9–e11. doi:10.1053/j.gastro.2019.01.031
- Schieppati A, Perico N, Remuzzi G. Tacrolimus and ciclosporin microemulsion in renal transplantation. Lancet. 2002;360(9335):799–800. doi:10.1016/S0140-6736(02)09908-7
- Morrison J, Ferguson E, Figueroa J, Karpen SJ. Features of cirrhotic cardiomyopathy early in the lives of infants with biliary atresia correlate with outcomes following kasai portoenterostomy, Hepatol commun. Hepatology Communications. 2022;6(6):1413–1424. doi:10.1002/hep4.1890
- Dai SY, Sun YQ, Wu Y, et al. Development and assessment of screening nomogram for biliary atresia based on hepatobiliary ultrasonographic features. Front Pediatr. 2021;9:625451. doi:10.3389/fped.2021.625451
- Murthy V, Altawallbeh G, Larson-Nath C, Karger AB, Thomas SN. Transient hyperphosphatasemia following pediatric liver transplantation in a patient with hepatic and skeletal abnormalities. Clin Chim Acta. 2021;519:48–50. doi:10.1016/j.cca.2021.03.030
- Harpavat S, Garcia-Prats JA, Anaya C, et al. Diagnostic yield of newborn screening for biliary atresia using direct or conjugated bilirubin measurements. JAMA. 2020;323(12):1141–1150. doi:10.1001/jama.2020.0837
- Wang J, Xu Y, Chen Z, et al. Liver Immune Profiling Reveals Pathogenesis and Therapeutics for Biliary Atresia, Cell. 2020;183:1867–1883. doi:10.1016/j.cell.2020.10.048
- Hamaguchi M, Sakamoto R, Kohrogi K, et al. Complete remission of refractory immunothrombocytopenic purpura after tacrolimus replacement with cyclosporine in a case of living related liver transplant. Exp Clin Transplant. 2021;19(11):1228–1231. doi:10.6002/ect.2021.0272
- Lacaille F. Liver transplantation and liver cell transplantation. Clin Res Hepatol Gastroenterol. 2012;36(3):304–307. doi:10.1016/j.clinre.2012.03.029
- Cross SA, Perry CM. Tacrolimus once-daily formulation: in the prophylaxis of transplant rejection in renal or liver allograft recipients. Drugs. 2007;67(13):1931–1943. doi:10.2165/00003495-200767130-00012
- Solari S, Cancino A, Wolff R, et al. Sublingual tacrolimus administration provides similar drug exposure to per-oral route employing lower doses in liver transplantation: a pilot study. Aliment Pharmacol Ther. 2017;45(9):1225–1231. doi:10.1111/apt.14022
- Florman S. Tacrolimus once-daily formulation in the prophylaxis of transplant rejection in renal or liver allograft recipients: a viewpoint by sander florman. Drugs. 2007;67(13):1944. doi:10.2165/00003495-200767130-00013
- Wang Q, Dai X, Yang W, et al. Caffeine protects against alcohol-induced liver fibrosis by dampening the cAMP/PKA/CREB pathway in rat hepatic stellate cells. Int Immunopharmacol. 2015;25(2):340–352. doi:10.1016/j.intimp.2015.02.012
- Anouti A, Patel MS, VanWagner LB, et al. An update on biliary atresia liver transplant candidate and recipient outcomes in the United States. Gastroenterology. 2023;164(6):S1238–S1239. doi:10.1016/S0016-5085(23)03875-1
- Sata N, Hirata Y, Lefor AK, Sata N. Long-term outcomes in pediatric patients who underwent living donor liver transplantation for biliary atresia. Surgery. 2022;171(6):1671–1676. doi:10.1016/j.surg.2021.11.027
- Serinet MO, Golmard JL, Jacquemin E, et al. Improving outcomes of biliary atresia: French national series 1986-2009. J Hepatol. 2013;58(6):1209–1217. doi:10.1016/j.jhep.2013.01.040
- Qiao G, Li L, Cheng W, Zhang Z, Ge J, Wang C. Conditional probability of survival in patients with biliary atresia after kasai portoenterostomy: a Chinese population-based study. J Pediatr Surg. 2015;50(8):1310–1315. doi:10.1016/j.jpedsurg.2015.03.062
- Dong R, Jiang J, Zhang S, Shen Z, Chen G. Development and validation of novel diagnostic models for biliary atresia in a large cohort of Chinese patients. EBioMedicine. 2018;34:223–230. doi:10.1016/j.ebiom.2018.07.025
- Davenport M, Stringer MD, Mieli-Vergani G, Kelly DA, McClean P, Spitz L. Seamless management of biliary atresia in England and Wales. Lancet. 2004;363(9418):1354–1357. doi:10.1016/S0140-6736(04)16045-5
- Tessier MEM, Cavallo L, Yeh J, et al. The stool microbiome may predict outcomes in infants with biliary atresia. Gastroenterology. 2019;156(6):S137. doi:10.1016/S0016-5085(19)37134-3
- Jain V, Burford C, Alexander EC, et al. Prognostic markers at adolescence in patients requiring liver transplantation for biliary atresia in adulthood. J Hepatol. 2019;71(1):71–77. doi:10.1016/j.jhep.2019.03.005
- Tucker RM, Feldman AG, Fenner EK, Mack CL. Regulatory T cells inhibit Th1 cell-mediated bile duct injury in murine biliary atresia. J Hepatol. 2013;59(4):790–796. doi:10.1016/j.jhep.2013.05.010
- Lee CS, Lin CR, Chua HH, et al. Gut bifidobacterium longum is associated with better native liver survival in patients with biliary atresia. JHEP Rep. 2024;6(7):101090. doi:10.1016/j.jhepr.2024.101090
- de Vries W, Hulscher JB, Hoekstra-Weebers JE, Houwen RH, Verkade HJ. Netherlands Study Group of Biliary Atresia Registry. Twenty-year transplant-free survival rate among patients with biliary atresia, Clin Gastroenterol Hepatol. 9. 2011. 1086–1091. 10.1016/j.cgh.2011.07.024
- Zarrinpar A, Busuttil RW. Immunomodulating options for liver transplant patients. Expert Rev Clin Immunol. 2012;8(6):565–578. doi:10.1586/eci.12.47
- Takeuchi H, Matsuno N, Senuma K, et al. Evidence of different pharmacokinetics including relationship among AUC, peak, and trough levels between cyclosporine and tacrolimus in renal transplant recipients using new pharmacokinetic parameter--why cyclosporine is monitored by C(2) level and tacrolimus by trough level. Biol Pharm Bull. 2008;31(1):90–94. doi:10.1248/bpb.31.90
- Keown PA. New concepts in cyclosporine monitoring. Curr Opin Nephrol Hypertens. 2002;11(6):619–626. doi:10.1097/00041552-200211000-00008
- Lilly LB, Grant D. Optimization of cyclosporine for liver transplantation. Transplant Proc. 2004;36(2):267S–270S. doi:10.1016/j.transproceed.2003.12.050
- Jorga A, Holt DW, Johnston A. Therapeutic drug monitoring of cyclosporine. Transplant Proc. 2004;36(2):396S–403S. doi:10.1016/j.transproceed.2004.01.013
- Oellerich M, Armstrong VW. Two-hour cyclosporine concentration determination: an appropriate tool to monitor neoral therapy? Ther Drug Monit. 2002;24(1):40–46. doi:10.1097/00007691-200202000-00008
- Shenoy S, Hardinger KL, Crippin J, et al. A randomized, prospective, pharmacoeconomic trial of neoral 2-hour postdose concentration monitoring versus tacrolimus trough concentration monitoring in de novo liver transplant recipients. Liver Transpl. 2008;14(2):173–180. doi:10.1002/lt.21355