Abstract
Objective and design
Reduced expression of histone deacetylase 2 (HDAC2) in alveolar macrophages and epithelial cells may account for reduced response of chronic obstructive pulmonary disease (COPD) patients to glucocorticoids. HDAC2 expression and its role in mediating glucocorticoid effects on fibroblast functions, however, has not been fully studied. This study was designed to investigate whether HDAC2 mediates glucocorticoid effects on release of inflammatory cytokines and matrix metalloproteinases (MMPs) from human lung fibroblasts.
Methods
Human lung fibroblasts (HFL-1 cells) were stimulated with interleukin (IL)-1 β plus tumor necrosis factor (TNF)-α in the presence or absence of the glucocorticoid budesonide. Cytokines (IL-6 and IL-8) were quantified by enzyme linked immunosorbent assay (ELISA) and MMPs (MMP-1 and MMP-3) by immunoblotting in culture medium. The role of HDAC2 was investigated using a pharmacologic inhibitor as well as a small interfering ribonucleic acid (siRNA) targeting HDAC2.
Results
We have demonstrated that budesonide concentration-dependently (10−10–10−7 M) inhibited IL-6, IL-8, MMP-1, and MMP-3 release by HFL-1 cells in response to IL-1β plus TNF-α. While an HDAC inhibitor significantly blocked the inhibitory effect of budesonide on human bronchial epithelial cells (HBECs) and monocytes (THP-1 cells), it did not block the inhibitory effect of budesonide on release of cytokines and MMPs from HFL-1 cells. Similarly, an HDAC2-siRNA blocked budesonide inhibition of cytokine release in HBECs, but it did not block the inhibitory effect of budesonide on HFL-1 cytokine and MMP release. Furthermore, budesonide significantly blocked release of cytokines and MMPs to a similar degree in normal and COPD lung fibroblasts as well as in HFL-1 cells exposed or not exposed to cigarette smoke extract.
Conclusion
These findings suggest that, in contrast to airway epithelial cells and monocytes/macrophages, HDAC2 is not required for budesonide to inhibit MMP and cytokine release by lung fibroblasts and this inhibitory pathway appears to be intact in cultured fibroblasts from COPD patients. These results also suggest that budesonide has the potential to modulate fibroblast-mediated tissue remodeling following airway inflammation in COPD, which is mediated via an HDAC2 independent pathway.
Keywords:
Introduction
Chronic obstructive pulmonary disease (COPD) is an increasing global health problem. Airway inflammation and tissue remodeling that leads to peribronchial fibrosis are characteristic of COPD, and contribute to the relentless progress of the disease.Citation12 Inhaled glucocorticoids are very effective anti-inflammatory agents in asthma, but have limited benefits in COPD. Reduced expression of histone deacetylase 2 (HDAC2) in alveolar macrophages and airway epithelial cells in COPD has been suggested to contribute to the limited ability of glucocorticoids to inhibit the inflammation present in COPD.Citation3,Citation4 However, HDAC2 expression in lung fibroblasts and its role in mediating the effect of glucocorticoids on lung fibroblast functions are unclear.
Histone acetyltransferases and histone deacetylases (HDACs) play important roles in regulating gene expression.Citation5 Acetylation of histone core structure results in opening up of the chromatin allowing transcription factors and ribonucleic acid (RNA) polymerase to bind to DNA and initiate gene transcription. Histone acetylation can be reversed by HDACs. There are eleven HDAC isoenzymes that deacetylate histones within the nucleus and specific HDACs may regulate different groups of genes.Citation6 Expression of inflammatory genes is regulated by acetylation of histone 4 and HDAC2.Citation7,Citation8 In COPD alveolar macrophages and airway biopsy specimens, histones that are associated with the promoter region of inflammatory genes are highly acetylated and the degree of acetylation increases with disease severity.Citation3 This increased acetylation of inflammatory genes seems to be not due to increased activity of acetyltransferase but rather due to the reduced expression of HDAC2 in human alveolar macrophages and lung tissue.Citation3
Lung fibroblasts play a key role in airway remodeling and tissue repair following inflammatory injury of small airways.Citation2 The expression of HDAC2 and its role in mediating glucocorticoid anti-inflammatory effect in fibroblasts has not been investigated. The current study, therefore, was designed to investigate HDAC2 expression in human lung fibroblasts from control and COPD subjects and to determine the role of HDAC2 in regulating the effect of budesonide on the release of inflammatory mediators by lung fibroblasts. To accomplish this, human lung fibroblasts were treated with interleukin (IL)-1β and tumor necrosis factor (TNF)-α, the two well-known pro-inflammatory cytokines that are believed to play a role in COPD and that stimulate human lung fibroblasts to synthesize and release several inflammatory mediators including IL-6 and IL-8, matrix metalloproteinase (MMP)-1 and MMP-3.Citation9–Citation11 The role of HDAC2 in mediating budesonide inhibition of the release of IL-6, IL-8, MMP-1, and MMP-3 was then assessed using a pharmacologic inhibitor and RNA interference.
Material and methods
Cell culture
Human fetal lung fibroblasts (HFL-1), obtained from the American Type Culture Collection (Manassas, VA, USA), were cultured with Dulbecco’s Modified Eagle’s Medium (DMEM; Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal calf serum (FCS) (Life Technologies), 50 U/mL penicillin, 50 μg/mL streptomycin (Life Technologies) and 1 μg/mL amphotericin B (X-Gen, Big Flats, NY, USA). The cells were cultured in 100-mm tissue culture dishes. Fibroblasts were trypsinized (0.05% trypsin, 0.481 mM ethylene diamine tetraacetic acid [EDTA]; Life Technologies), plated into tissue culture plates, grown to near confluence and used for assays.
Normal human bronchial epithelial cells (HBECs) were acquired from bronchial biopsies under a research protocol approved by the University of Nebraska Institutional Review Board using a previously published method with slight modifications.Citation12 Briefly, the biopsy tissues were allowed to attach to the type I collagen-coated 60 mm tissue culture dishes under serum-free conditions using a 1:1 mixture of LHC-9/RPMI-1640 (Laboratory of Human Carcinogenesis-9/Roswell Park Memorial Institute-1640) medium (Life Technologies) without addition of cortisol.Citation13 Medium was changed every other day. After 7–10 days culture, HBECs were trypsinized and plated on collagen (PureCol; Advanced BioMatrix, San Diego, CA, USA) coated tissue culture dishes (Falcon; Becton-Dickinson Labware, Lincoln Park, NJ, USA) at 37°C in a humidified, 5% CO2 atmosphere. Cells were then passaged once a week at a 1:3 ratio. Cells between the 3rd and 10th passage were characterized by staining for pancytokeratin and vimentin and used for experiments.
Monocytic cell line cells (THP-1), obtained from the American Type Culture Collection, were cultured in RPMI-1640 supplemented with 10% FCS (Life Technologies).
Primary human lung fibroblasts were cultured from lung tissues obtained from subjects with or without clinical and functional signs of COPD as described earlier.Citation14 All subjects were undergoing surgery and acquisition of samples was approved by the Human Studies Committee of the Medical Board of the State of Schleswig-Holstein (Germany). All subjects also provided written informed consent for research. Fibroblasts from six normal subjects (forced expiratory volume in 1 second [FEV1]: 89.4%±5.7% predicted [mean + standard error of the mean], range 75.6%–115.4%) and six COPD subjects (FEV1: 57.0%±1.6% predicted [mean ± standard error of the mean], range 53.2%–63.1%) were used in the current study.
Cytokine quantification by ELISA
IL-6 was quantified by enzyme linked immunosorbent assay (ELISA) as described previouslyCitation15 Briefly, 96-well plates were coated with capturing antibodies specific for IL-6 (R&D Systems, Minneapolis, MN, USA) at 4°C overnight. Samples and standard (R&D Systems) were added and allowed to react for 1 hour. After washing, the second antibody (Calbiochem, EMD Millipore, Billerica, MA, USA) was applied and incubated for 1 hour at room temperature. After washing, horseradish peroxidase (HRP)-conjugated anti-rabbit antibody (Rockland Immunochemicals, Gilbertsville, PA, USA) was applied for 1 hour at room temperature. O-phenylenediamine (OPD) substrate was applied and absorbance read at 495 nm wavelength.
IL-8 was quantified by using Human IL-8 DuoSet Kit (R&D Systems) following the manufacturer’s instruction.
Immunoblotting
To evaluate MMP-1 and -3, conditioned media (2 mL) collected from the monolayer culture of the fibroblasts were concentrated by ethanol precipitation and re-suspended in distilled H2O (50 μL).Citation16 The samples (15 μL) were then added to the same volume of 2x concentrated electrophoresis sample buffer (0.5 M Tris pH 6.8, 2% sodium dodecyl sulfate [SDS], 20% glycerol, 0.1 % bromophenol blue) and heated for 5 minutes at 95°C. As MMPs were evaluated in culture medium in which there are no reference proteins, care was taken to load equal volumes and quantitative results are expressed relative to control cells treated only with cytokines (IL-1β and TNF-α). Thirty microliters of each sample was loaded into each lane of a 10% SDS-polyacrylamide gel electrophoresis (PAGE) gel and electrophoresed. The proteins were transferred to a polyvinylidene difluoride (PVDF) membrane with a semi-dry transfer apparatus. The membrane was blocked in 5% milk in phosphate buffered saline (PBS)-Tween at room temperature for 1 hour, then exposed to primary antibodies (mouse anti-human MMP-1 or MMP-3 from R&D Systems), which were subsequently detected using HRP-conjugated rabbit anti-mouse immunoglobulin G (IgG) in conjunction with an enhanced chemiluminescence (ECL) detection system.
For HDAC, whole cell lysate proteins were extracted with 100 μL per 60 mm dish of cell lysis buffer (35 mM Tris-HCl, pH 7.4, 0.1% Triton X-100, 0.4 mM ethylene glycol tetraacetic acid [EGTA], 10 mM MgCl2) containing a protease inhibitor cocktail (Sigma-Aldrich, St Louis, MO, USA). Cells were briefly sonicated followed by centrifugation at 10,000g for 10 minutes at 4°C and the supernatant was subjected to immunoblotting for HDAC2 (1:200 dilution; Abcam, Cambridge, MA, USA), with β-actin (1:4000 dilution; Sigma-Aldrich) as loading control. SDS-PAGE electrophoresis, protein transfer and immunoblotting were conducted as described above.
HDAC inhibition
To inhibit HDAC, a non-selective HDAC inhibitor, trichostatin A (TSA; Cell Signaling, Danvers, MA, USA) was used. After incubation of HFL-1, HBECs and THP-1 cells with various concentrations of TSA for 30 minutes, varying concentrations of budesonide (AstraZeneca, Lund Sweden) and IL-1β plus TNF-α (R&D Systems) were added to the final concentrations of IL-1β plus TNF-α, 1 ng/mL each. After 24 hours, media were harvested for quantification of IL-6 and IL-8 by ELISA, as well as MMP-1 and MMP-3 by immunoblotting. Cells were trypsinized and counted with a Coulter Counter (Beckman Coulter, Brea, CA, USA). Levels of the cytokines were normalized by cell number for each experiment.
Selective inhibition of HDAC2 by RNA interference
To selectively silence HDAC2, RNA interference was performed. Briefly, cells were seeded in 6-well dishes at a cell density of 2 × 105 cells per well. The next day, cells were transfected with small interfering (si)RNA targeting HDAC2 or nontargeting control siRNA (final concentration of siRNA was 50 nM; Santa Cruz Biotechnology Inc, Santa Cruz, CA, USA) in Opti-MEM (Invitrogen, Carlsbad CA, USA) using Lipofectamine 2000 (Invitrogen). After 16 hours transfection, media were changed to 10% FCS-DMEM for HFL-1 cells or LHC-9/RPMI for HBECs. After 24 hours, the cells were treated with cytokines (IL-1β and TNF-α) and/or budesonide (AstraZeneca). Media were harvested and the cell lysates were extracted on day 4 and the efficacy of RNA interference was assessed by immunoblotting.
Preparation of cigarette smoke extract (CSE)
CSE was prepared with a modification of the method reported previouslyCitation17 Briefly, the smoke from one 84 mm cigarette (research cigarette 3R4F; University of Kentucky, Lexington, KY, USA) without filter was bubbled through 15 mL of deionized water at a speed of 50 cc/minute. The filtered solution with a 0.22 μm pore filter (Lida Manufacturing, Kenosha, WI, USA) was considered to be 100% CSE and applied to fibroblast cultures within 30 minutes of preparation. In the current study, fibroblasts were exposed to 5% CSE diluted with serum free DMEM.
Statistical analysis
Results were always confirmed by repeating each experiment on separate occasions at least three times. Statistical comparisons were based on separate experiments. Group data were analyzed by one-way analysis of variances (ANOVA) followed by Tukey’s test or two-way ANOVA followed by Bonferroni test using the GraphPad Prism 4 software (GraphPad Software, Inc., La Jolla, CA, USA). P < 0.05 was considered significant.
Results
HDAC inhibitor blocked budesonide effect in HBECs but not in HFL-1 cells
In order to study the effect of budesonide on release of cytokines and MMPs by fibroblasts, HFL-1 cells were treated with IL-1β plus TNF-α (1 ng/mL, each) in monolayer culture to assess IL-6 and IL-8 as well as MMP-1 and MMP-3. As expected IL-1β plus TNF-α significantly stimulated IL-6, IL-8, MMP-1, and MMP-3 release by HFL-1 cells (–). Budesonide (from 10−10 to 10−7 M) significantly inhibited IL-1β plus TNF-α-induced release of all four mediators (–, P < 0.01). TSA (10−9–10−7 M), a nonselective HDAC inhibitor, did not block budesonide inhibition of cytokine or MMP release (–) in human lung fibroblasts. Interestingly, in the presence of IL-1β plus TNF-α, TSA (10−7 M) significantly inhibited MMP-1 release (), but significantly stimulated MMP-3 release ().
Figure 1 Budesonide inhibits release of cytokines and MMPs by human lung fibroblasts.
Notes: HFL-1 cells were cultured until sub-confluence and treated with varying concentrations of budesonide for 30 minutes followed by stimulation with cytokines (IL-1β plus TNF-α, 1 ng/ml each). After 24 hours, medium was harvested for quantification of IL-6 (A) and IL-8 (B) by ELISA, and MMP-1 (C) and MMP-3 (D) by immunoblotting. (A and B): Effect on IL-6 and IL-8 release. Cell number was counted with a Coulter counter and the cytokine level was normalized to the cell number. Vertical axis: amount of IL-6 or IL-8 expressed as percentage of response in cytokines-only treated (IL-1β plus TNF-α) cells; horizontal axis: treatment with cytokines and varying concentrations of budesonide. Data presented are the mean ± SEM of three separate experiments. (C and D): Inset is an example of immunoblot image. In order to make quantitative comparisons, the same volume of media from each condition was harvested and precipitated for loading. Bar graphs presented are densitometric analysis of immunoblots expressed as the mean ± SEM of three separate experiments. Vertical axis: density of the image expressed as percentage of density in the cytokines-only treated group; horizontal axis: treatment with cytokines and varying concentrations of budesonide. *P < 0.05; **P < 0.01 compared to cytokines alone by one way analysis of variance followed by Tukey’s test.
Abbreviations: ELISA, enzyme linked immunosorbent assay; IL, interleukin; MMP, matrix metalloproteinase; SEM, standard error of the mean; TNF, tumor necrosis factor.
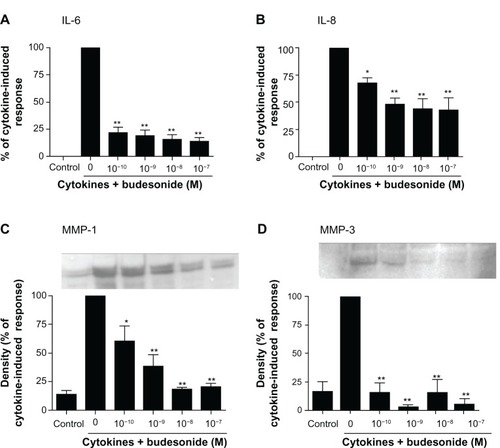
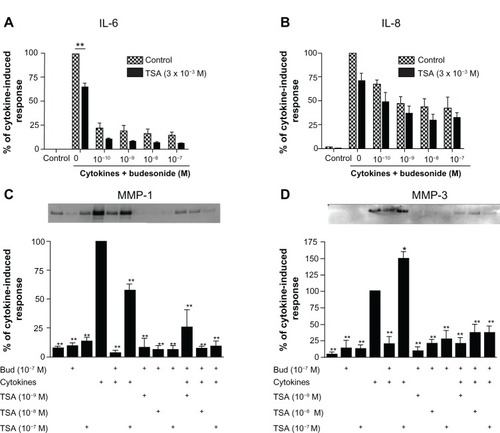
Figure 2 Trichostatin A does not block the budesonide effect in human lung fibroblasts.
Notes: HFL-1 cells were plated and grown to sub-confluence. Cells were then rinsed with DMEM and treated with TSA for 30 minutes. Following this, varying concentrations of budesonide and IL-1β plus TNF-α were added such that the final concentrations of IL-1β and TNF-α were 1 ng/mL each and the concentrations of budesonide were as described in the figure. After 24 hours, media were harvested for quantification of IL-6 (A), IL-8 (B), and MMP-1 and -3 (C and D). (A and B) Effect on IL-6 and IL-8 release (ELISA). Cell number was counted with a Coulter Counter and the cytokine level was normalized to the cell number. Vertical axis: amount of IL-6 or IL-8 expressed as percentage of response in the cytokines-only treated group; horizontal axis: treatment with cytokines and varying concentrations of budesonide. hatched bars: control (cells without TSA treatment); solid bars: cells treated with TSA (3 × 10−8 M). Data presented are the mean ± SEM of three separate experiments. **P < 0.01 by two way analysis of variance followed by Bonferroni post-hoc test. (C and D) Densitometric analysis of the MMP-1 and -3 immunoblots (insets). In order to make quantitative comparisons, the same volume of media from each condition was harvested and precipitated for loading. Vertical axis: density expressed as percentage of the cytokines-only treated group. horizontal axis: treatment with cytokines, budesonide (10−7 M) and varying concentrations of TSA. Data presented are the mean ± SEM of three separate experiments. *P < 0.05; **P < 0.01 compared to cytokine alone by two way analysis of variance followed by Bonferroni test.
Abbreviations: Bud, budesonide; DMEM, Dulbecco’s Modified Eagle’s Medium; ELISA, enzyme linked immunosorbent assay; HFL, human lung fibroblast; IL, interleukin; MMP, matrix metalloproteinase; SEM, standard error of the mean; TNF, tumor necrosis factor; TSA, trichostatin A.
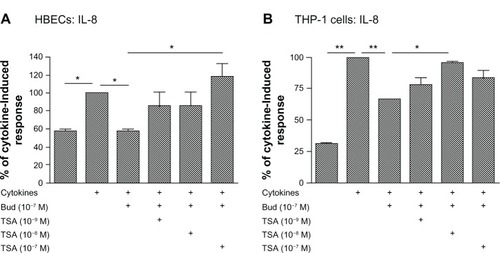
In contrast to the lack of effect of TSA on budesonide inhibition of mediator release in fibroblasts, TSA significantly blocked the inhibitory effect of budesonide (10−7 M) on IL-8 release in HBECs (, TSA at 10−7 M) and in the THP-1 cell line (, TSA at 10−8 M). Similarly, TSA also significantly blocked the inhibitory effect of budesonide on cytokine-induced IL-6 release by HBECs; budesonide (10−7 M) reduced release to 59.0% ± 9.5% (mean ± standard error of the mean) of cytokine-stimulated cells (100%) and TSA blocked this effect resulting in 98.2% ± 10.2% release (mean ± standard error of the mean; control non-stimulated cells release was 45.7% ± 2.9% of cytokine-stimulated cells). These results demonstrate that budesonide can inhibit fibroblast mediator release via an HDAC independent mechanism, while the effect on other cells requires HDAC.
Figure 3 Trichostatin A blocks budesonide effect in HBECs (A) and monocytes (THP-1; B).
Notes: Nearly confluent HBECs were pretreated with varying concentrations of TSA for 30 minutes. Cells were then treated with budesonide (10−7 M) in the presence or absence of cytokines IL-1b plus TNF-α (1 ng/ml each). After 24 hours, medium was harvested for quantification of IL-8 by ELISA. Cell number was counted with a Coulter Counter and the cytokine level was normalized to the cell number. Vertical axis: IL-8 amount expressed as percent of response in cytokines-only treated cells; horizontal axis: treatment. Data presented are the mean ± SEM of three separate experiments. *P < 0.05; **P < 0.01 by one way analysis of variance followed by Tukey’s testAbbreviations: Bud, budesonide; ELISA, enzyme linked immunosorbent assay; HBECs, human bronchial epithelial cells; IL, interleukin; SEM, standard error of the mean; TNF, tumor necrosis factor; TSA, trichostatin A.
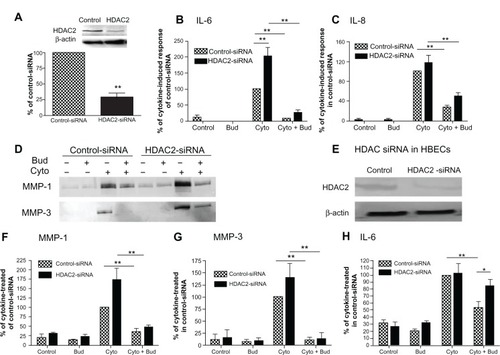
HDAC2 suppression by siRNA blocked the effect of budesonide in HBECs but not in HFL-1 cells
In order to further investigate the role of HDAC in mediating budesonide inhibition of cytokine and MMP release by fibroblasts, HFL-1 cells were transfected with an siRNA specifically targeting HDAC2, followed by treatment with budesonide. HDAC2 was significantly suppressed by the siRNA (; P < 0.01), however, despite suppression of HDAC2, budesonide (10−7 M) still significantly inhibited cytokine-stimulated IL-6, IL-8, MMP-1, and MMP-3 (–, P < 0.01). In order to ensure that the siRNA was active, the effect of siRNA suppression of HDAC2 was investigated on budesonide (5 × 10−7 M) inhibition of IL-6 release by HBECs. The siRNA targeting HDAC2 was found to significantly abolish the effect of the high concentration of budesonide in HBECs (, P < 0.05).
Figure 4 HDAC2 suppression by siRNA and its role in mediating the effect of budesonide on human lung fibroblasts and bronchial epithelial cells.
Notes: HFL-1 cells or HBECs were transfected with either a negative control siRNA or an siRNA targeting HDAC2 (HDAC2-siRNA). Cells were then treated with cytokines IL-1β plus TNF-α (1 ng/mL each) in the presence or absence of budesonide (10−7 M). Hatched bars: cells transfected with non-targeting control-siRNA; solid bars: cells transfected with siRNA specifically targeting HDAC2. (A) Suppression of HDAC2 by siRNA in HFL-1 cells. Density of HDAC2 was normalized by β-actin density for each experiment, and then data were expressed a mean ratio (± SEM) of HDAC2-siRNA density versus control-siRNA density in three separate experiments. Inset: Image of immunoblot from one representative experiment. Top band: HDAC2; bottom band: β-actin. **P < 0.01 compared to control-siRNA. (B and C) Effect of HDAC2 suppression by siRNA on budesonide (10−7 M) inhibition of IL-6 and IL-8 release (ELISA) by HFL-1 cells (data were normalized to the cell number; data are shown as the mean ± SEM of three separate experiments). (D, F and G) Effect of HDAC2 suppression by siRNA on budesonide (10−7 M) inhibition of MMP-1 and MMP-3 release (immunoblotting) by HFL-1 cells. Data presented are one representative of three separate experiments (D) or the mean ± SEM of three separate experiments. In order to make quantitative comparisons, the same volume of media from each condition was harvested and precipitated for loading. (E and H) Effect of HDAC2 suppression by siRNA on budesonide (5 × 10−7 M) inhibition of IL-6 release by HBECs. After siRNA transfection, cells were treated with cytokines and budesonide as indicated for 24 hours. Media were harvested and IL-6 was quantified by ELISA (data were normalized to the cell number). (E) Immunoblots of HDAC2 and β-actin, showing suppression of HDAC2 by HDAC2-siRNA but not by control-siRNA. (H) Vertical axis: IL-6 amount expressed as percent of cytokine in control-siRNA treated cells; horizontal axis: treatment. Data presented are the mean ± SEM of two different HBEC cell lines. *P < 0.05, **P < 0.01.
Abbreviations: Bud, budesonide; Cyto, cytokines; ELISA, enzyme linked immunosorbent assay; HBEC, human bronchial epithelial cells; HDAC, histone deacetylase; HFL, human lung fibroblast; IL, interleukin; MMP, matrix metalloproteinase; SEM, standard error of the mean; si, small interfering; TNF, tumor necrosis factor; TSA, trichostatin A.
Budesonide effect was not impaired in COPD lung fibroblasts and in HFL-1 cells exposed to cigarette smoke
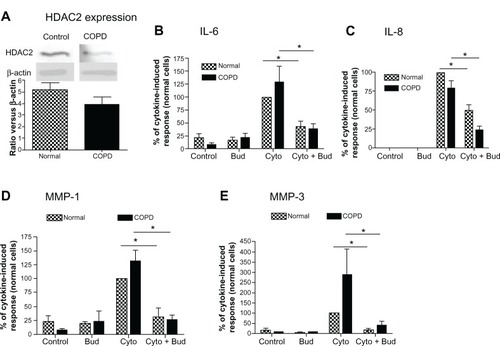
Finally, since HDAC2 expression has been reported to be reduced in alveolar macrophages from COPD patients,Citation3 HDAC2 protein expression was examined in lung fibroblasts cultured from COPD patients and compared to that in fibroblasts obtained from normal control subjects. While the expression of HDAC2 was numerically lower in COPD fibroblasts than in normal lung fibroblasts, the difference was not statistically significant (; 24% reduction, 4.0 ± 1.5 versus 5.2 ± 1.4 [mean ± standard error of the mean, N = 6]). Furthermore, budesonide at low concentration (10−9M) inhibited cytokine-induced IL-6, IL-8, MMP-1 and MMP-3 from both normal and COPD fibroblasts to a similar degree (–; P < 0.05).
Figure 5 HDAC expression in the lung fibroblasts from COPD patients and its role in mediating budesonide effect.
Notes: (A) HDAC2 expression in the fibroblasts isolated from normal subjects and COPD patients. Density of HDAC2 was normalized by β-actin density for each experiment, and then data were expressed as mean + SEM of six separate isolations of primary fibroblasts from each group. Inset: Image of representative immunoblot Top band: HDAC2; bottom band: β-actin. (B–E) effect of budesonide on IL-6 (B), IL-8 (C), MMP-1 (D), and MMP-3 (E) release by lung fibroblasts from normal subjects and COPD patients under control conditions and after stimulation with cytokines IL-1 β plus TNF-α (1 ng/ml each). IL-6 and IL-8 were quantified by ELISA (data were normalized to the cell number), and MMP-1 and MMP-3 by immunoblotting. In order to make quantitative comparisons, the same volume of media from each condition was harvested and precipitated for loading. Vertical axis: mediator amount expressed as percent of response in cytokines-only treated fibroblasts from normal subjects; horizontal axis: treatment. hatched bars: fibroblasts from normal subjects; solid bars: fibroblasts from COPD patients. *P < 0.05. Data presented are the mean + SEM of six separate primary cells from each group.
Abbreviations: Bud, budesonide; COPD, chronic obstructive pulmonary disease; Cyto, cytokines; ELISA, enzyme linked immunosorbent assay; HDAC, histone deacetylase; IL, interleukin; MMP, matrix metalloproteinase; SEM, standard error of the mean; TNF, tumor necrosis factor.
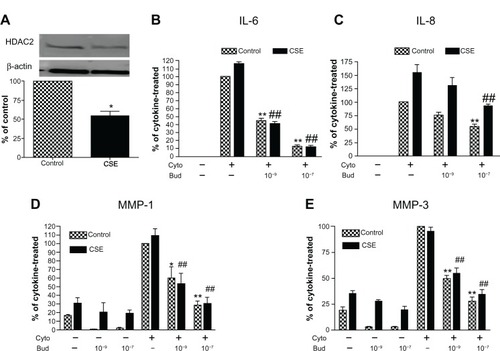
Interestingly, HDAC2 protein was significantly decreased to 54.0% ± 6.1% in the HFL-1 cells exposed to 5% CSE for 72 hours compared to the cells without CSE exposure (, P < 0.05). Consistent with previous reports,Citation18,Citation19 CSE stimulated IL-6 and IL-8 release by human lung fibroblasts under control conditions as well as release of MMP-1. Cytokines (IL-1β plus TNF-α) markedly stimulated IL-6, IL-8, MMP-1, and MMP-3 in both CSE-pretreated and control fibroblasts. Budesonide at concentrations of 10−9 and/or 1 0−7 M significantly inhibited cytokine-induced IL-6, IL-8, MMP-1, and MMP-3 from both control and CSE-pre-exposed fibroblasts to a similar degree, suggesting that any inhibition of HDAC2 that may result from smoke exposure did not alter the ability of budesonide to inhibit mediator release (–).
Figure 6 CSE effect on HDAC expression by human lung fibroblasts and its role in mediating budesonide effect.
Notes: (A) Effect of CSE on HDAC2 expression. HFL-1 cells were treated with 5% CSE for 72 hours. HDAC2 level was assessed by immunoblotting. Density of HDAC2 was normalized by β-actin density for each experiment, and then data were expressed as an average ratio of HDAC2 density in CSE-exposed fibroblasts versus HDAC2 density in control fibroblast. Inset Image of immunoblot Top band: HDAC2; bottom band: β-actin. Image data is one representative of three separate experiments and bar graph shows the mean ± SEM of three separate experiments. (B–E) Budesonide effect on IL-6 (B), IL-8 (C), MMP-1 (D), and MMP-3 (E) release induced by cytokines IL-1 β plus TNF-α (1 ng/ml each) in the cells exposed or not exposed (control) to CSE. HFL-1 cells were pre-treated with or without 5% CSE for 72 hours. Cells were then stimulated with cytokines in the presence of varying concentrations of budesonide. Vertical axis: protein levels expressed as percent of response in cytokines-only treated control fibroblasts; horizontal axis: treatment. Hatched bars: without CSE exposure; solid bars: cells exposed to 5% CSE. Data presented are the mean ± SEM of three separate experiments. *P < 0.05; **P < 0.01 compared to cytokines-only treated control fibroblasts; ##P < 0.01 compared to cytokines-only treated fibroblast exposed to CSE.
Abbreviations: Bud, budesonide; CSE, cigarette smoke extract; Cyto, cytokines; HDAC, histone deacetylase; HFL, human lung fibroblast; IL, interleukin; MMP, matrix metalloproteinase; SEM, standard error of the mean; TNF, tumor necrosis factor.
Discussion
The current study demonstrates that budesonide inhibits release of cytokines (IL-6 and IL-8) and MMPs (MMP-1 and MMP-3) from IL-1β plus TNF-α-stimulated human lung fibroblasts (HFL-1 and primary cells), HBECs, and cells from a human monocytic cell line (THP-1). Human lung fibroblasts express HDAC2, but neither inhibition of HDAC by a pharmacologic inhibitor TSA nor suppression with HDAC2 specific siRNA affected budesonide inhibition of mediator release from fibroblasts. This contrasts with HBECs and THP-1 cells, in which the inhibitory effect of budesonide was significantly blocked by HDAC inhibition. HDAC2 protein expression in lung fibroblasts from COPD patients was slightly but not significantly lower than that in normal lung fibroblasts whereas it was significantly decreased by nearly 50% in the lung fibroblasts (HFL-1) exposed to CSE compared to nonexposed cells. Nevertheless, budesonide inhibited production of IL-6, IL-8, MMP-1, and MMP-3 in both normal and COPD lung fibroblasts or CSE-exposed and nonexposed cells similarly. Taken together, these results demonstrate that budesonide can inhibit fibroblast mediator release via an HDAC independent mechanism and that fibroblasts from COPD patients or CSE-exposed fibroblasts are sensitive to this inhibition.
Histone acetylation and deacetylation play an important role in gene expression. Acetylation of core histones by histone acetyltransferase results in opening the chromatin structure to allow transcription factors and RNA polymerase binding to DNA thus, initiating gene transcription.Citation5 Histone acetylation is reversed by HDACs, and by this mechanism gene expression is turned off. There are 11 HDAC isoenzymes described and, of these, HDAC2 has been reported to be critically important in mediating glucocorticoid suppression of airway inflammation.Citation4,Citation8 Budesonide is a glucocorticoid with potent anti-inflammatory effects and limited systemic bioactivity due to extensive (90%) first-pass hepatic metabolism by cytochrome P450 enzymes.Citation20 Inhalation of glucocorticoids has been shown to be effective in suppressing airway inflammation in asthmatics but is generally less effective in COPD. Reduction of HDAC2 expression in airway macrophages and epithelial cells in COPD patients is believed to account for the inflammation in COPD being less responsive to glucocorticoid therapy.Citation3 This resistance poses a paradox, as glucocorticoids have well-demonstrated therapeutic benefits in COPD, including improvement in airflow and reduction in the frequency of exacerbations. Because the clinical benefits develop slowly over a matter of weeks, an anti-inflammatory action has been suggested, despite the reports that macrophages and epithelial cells are resistant to the anti-inflammatory effects of glucocorticoids in COPD.Citation3,Citation4,Citation8
Consistent with previous reports, the current study demonstrated that budesonide inhibited IL-6 and IL-8 release by human bronchial epithelial cells,Citation21 lung fibroblasts,Citation11 and monocytic cells.Citation22 Moreover, also consistent with previous reports,Citation23,Citation24 inhibition of HDAC activity by TSA blocked the inhibitory effect of budesonide on IL-8 and IL-6 release by HBECs and THP-1 cells. In contrast, the inhibitory effect of budesonide on the lung fibroblasts was not sensitive to TSA. Interestingly, in the presence of IL-1β plus TNF-α, TSA alone inhibited MMP-1 release but stimulated MMP-3 release indicating that it had an effect on the fibroblasts, although it did not block budesonide-mediated inhibition. Consistent with this, TSA has been reported to up-regulate MMP-11 expression in colon cancer cells,Citation25 but TSA inhibited IL-1β-stimulated MMP-1, MMP-3, and MMP-13 in human articular chondrocytes.Citation26 The mechanisms for TSA stimulation or inhibition of MMPs, however, remains to be determined. The current study, however, confirms previous reports that histone deacetylation plays a key role in the glucocorticoid-mediated inhibition of mediator release by HBECs and monocytes/macrophages, and demonstrates that glucocorticoids may still modulate the inflammatory response by inhibiting the release of mediators from lung fibroblasts, which is not HDAC dependent.
TSA is not specific for HDAC2 and a specific HDAC2 inhibitor was not available. However, we also used siRNA knockdown to specifically target HDAC2. This approach had a clear effect in blocking budesonide-mediated inhibition of mediator release in HBECs, but had no effect in lung fibroblasts. siRNA knockdown was confirmed by western blotting. As is usually the case, knockdown was not complete in either cell type, although HDAC2 expression was reduced by 75% in lung fibroblasts and was similarly reduced in HBECs when normalized to β-actin. Our experiments cannot exclude the possibility of an effect of residual HDAC2 in fibroblasts, and it is possible that the amount of HDAC2 activity required may vary among cell types. Importantly, however, the results with the siRNA and the TSA consistently support no role for HDAC2 in mediating the effect of budesonide in lung fibroblasts.
Fibroblasts are one of the major cell types present in the lung. They are also the major cell type present in the subepithelial tissue in the airways and comprise up to 25% of alveolar cells, although the markedly attenuated structure of the alveolus makes them difficult to recognize on routine histology.Citation27 In both locations, fibroblasts are believed to be the major source of extracellular matrix macromolecules. Fibroblasts are also potent sources of other mediators. These include growth factors for epithelial and endothelial cells, and enzymes that can mediate the degradation of connective tissue. Fibroblasts also produce both inflammatory mediators and enzymes that can modulate the inflammatory response. The relative importance of various cell types in driving inflammation is not delineated. However, fibroblasts, because of their presence in large numbers in normal tissues, may be particularly important in mediating chronic inflammatory processes. In this context, fibroblasts respond vigorously to the early inflammatory response mediators IL-1β and TNF-α.Citation28,Citation29 These mediators induce a number of responses in fibroblasts including release of the cytokines IL-6,Citation10 IL-8,Citation30 and MMPs.Citation16 Thus, fibroblasts may participate in the amplification of IL-1β and TNF-α-induced inflammation. The quantitative production of mediators by fibroblasts may be less than that of monocytes/macrophages or epithelial cells, however, this does not exclude a role for fibroblasts in amplifying and sustaining an inflammatory response. The current study provides evidence that the mechanism of glucocorticoid inhibition of lung fibroblast-mediated inflammation differs from that of monocytes/macrophages and airway epithelial cells, which may help to explain the clinical benefits of inhaled glucocorticoids in COPD.
We also examined expression of HDAC2 in the lung fibroblasts from normal and COPD subjects as well as in HFL-1 cells exposed to 5% CSE for 72 hours. In contrast to the reduced HDAC2 expression reported in alveolar macrophages from COPD patients,Citation3 there was no statistically significant difference in HDAC2 expression between normal and COPD lung fibroblasts. Nevertheless, the slight numerical difference observed (24% reduction in COPD lung fibroblasts, N = 6) in the current study does not exclude the possibility that evaluation of a larger number of samples, or samples from more severely affected COPD patients, might show a statistically significant difference. Based on the data obtained a study of 25 subjects per group would be required to have an 80% chance of showing a statistically significant difference of the size observed in the current study. Thus, a modest inhibition of HDAC2 in lung fibroblasts from COPD patients cannot be excluded. In support of this concept, CSE exposure resulted in significant decline of HDAC2 expression in HFL-1 cells. However, in both the normal and COPD fibroblasts as well as in HFL-1 cells exposed or not exposed to CSE, the release of IL-6, IL-8, MMP-1, and MMP-3 in response to IL-1β and TNF-α was significantly inhibited by budesonide to a similar degree. Thus, the reduction of HDAC activity appeared to have no effect on glucocorticoid inhibition of mediator release from lung fibroblasts, further supporting our finding that in human lung fibroblasts budesonide inhibition of inflammatory mediator release is independent of HDAC2 level.
Conclusion
HDAC2 is expressed in human lung fibroblasts. Budesonide inhibits release of IL-6, IL-8, MMP-1, and MMP-3 from human lung fibroblasts stimulated by IL-1β plus TNFα. Suppression of HDAC2 by either a pharmacologic inhibitor (TSA) or an siRNA targeting HDAC2 does not block the inhibitory effects of budesonide in fibroblasts, which contrasts with HDAC-dependent inhibition in HBECs and monocytic cells. The demonstration that the anti-inflammatory effects of budesonide in lung fibroblasts are HDAC-independent suggests that budesonide has the potential to modulate fibroblast-mediated tissue remodeling following airway inflammation in COPD and provides a mechanism for the therapeutic benefits of glucocorticoids in COPD.
Acknowledgments
The authors are grateful for the secretarial support of Ms Lillian Richards. This work was supported by AstraZeneca and by the Larson Endowment, University of Nebraska Medical Center.
Disclosure
AM-L is an employee of AstraZeneca. EW was an employee of AstraZeneca at the time when the study was performed and the manuscript written. The authors report no other conflicts of interest.
References
- HoggJCPathophysiology of airflow limitation in chronic obstructive pulmonary diseaseLancet2004364943570972115325838
- SpurzemJRRennardSIPathogenesis of COPDSemin Respir Crit Care Med200526214215316088433
- ItoKItoMElliottWMDecreased histone deacetylase activity in chronic obstructive pulmonary diseaseN Engl J Med2005352191967197615888697
- BarnesPJRole of HDAC2 in the pathophysiology of COPDAnnu Rev Physiol20097145146418817512
- AdcockIMItoKBarnesPJHistone deacetylation: an important mechanism in inflammatory lung diseasesCOPD20052444545517147010
- ThiagalingamSChengKHLeeHJMinevaNThiagalingamAPonteJFHistone deacetylases: unique players in shaping the epigenetic histone codeAnn N Y Acad Sci20039838410012724214
- ItoKBarnesPJAdcockIMGlucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12Mol Cell Biol200020186891690310958685
- BarnesPJHistone deacetylase-2 and airway diseaseTher Adv Respir Dis20093523524319812111
- ZhuYKLiuXDSkoldCMSynergistic neutrophil elastase-cytokine interaction degrades collagen in three-dimensional cultureAm J Physiol Lung Cell Mol Physiol20012814L868L87811557590
- HarigaiMHaraMKitaniAInterleukin-1 and tumor necrosis factor-alpha synergistically increase the production of interleukin 6 in human synovial fibroblastJ Clin Lab Immunol19913431071131667942
- SpoelstraFMPostmaDSHovengaHNoordhoekJAKauffmanHFAdditive anti-inflammatory effect of formoterol and budesonide on human lung fibroblastsThorax200257323724111867828
- KelsenSGMardiniIAZhouSBenovicJLHigginsNCA technique to harvest viable tracheobronchial epithelial cells from living human donorsAm J Respir Cell Mol Biol19927166721320903
- BeckmannJDTakizawaHRombergerDSerum-free culture of fractionated bovine bronchial epithelial cellsIn Vitro Cell Dev Biol199228A139461730570
- HolzOZuhlkeIJaksztatELung fibroblasts from patients with emphysema show a reduced proliferation rate in cultureEur Respir J200424457557915459135
- StrizIMioTAdachiYBazilVRennardSThe CD14 molecule participates in regulation of IL-8 and IL-6 release by bronchial epithelial cellsImmunol Lett19986231771819698117
- KamioKLiuXDSugiuraHStatins inhibit matrix metalloproteinase release from human lung fibroblastsEur Respir J201035363764619797126
- CarpHJanoffAPossible mechanisms of emphysema in smokers. In vitro suppression of serum elastase-inhibitory capacity by fresh cigarette smoke and its prevention by antioxidantsAm Rev Respir Dis19781183617621101105
- ShizuMItohYSunaharaRCigarette smoke condensate upregulates the gene and protein expression of proinflammatory cytokines in human fibroblast-like synoviocyte lineJ Interferon Cytokine Res200828850952118729741
- NumanamiHKoyamaSNelsonDKSerine protease inhibitors modulate smoke-induced chemokine release from human lung fibroblastsAm J Respir Cell Mol Biol200329561361912738688
- AnderssonPRyrfeldtABiotransformation of the topical glucocorticoids budesonide and beclomethasone 17 alpha, 21-dipropionate in human liver and lung homogenateJ Pharm Pharmacol198436117637656150981
- StrandbergKPalmbergLLarssonKEffect of budesonide and formoterol on IL-6 and IL-8 release from primary bronchial epithelial cellsJ Asthma200845320120318415826
- LarssonSLofdahlCGLindenMIL-2 and IL-4 counteract budesonide inhibition of GM-CSF and IL-10, but not of IL-8, IL-12 or TNF-alpha production by human mononuclear blood cellsBr J Pharmacol1999127498098610433506
- ChenYWatsonAMWilliamsonCDGlucocorticoid receptor and histone deacetylase-2 mediate dexamethasone-induced repression of MUC5AC gene expressionAm J Respir Cell Mol Biol201247563764422798432
- TsaprouniLGItoKAdcockIMPunchardNSuppression of lipopolysaccharide- and tumour necrosis factor-alpha-induced interleukin (IL)-8 expression by glucocorticoids involves changes in IL-8 promoter acetylationClin Exp Immunol2007150115115717711487
- BarrasaJIOlmoNSantiago-GomezAHistone deacetylase inhibitors upregulate MMP11 gene expression through Sp1/Smad complexes in human colon adenocarcinoma cellsBiochim Biophys Acta20121823257058122227581
- WangXSongYJacobiJLTuanRSInhibition of histone deacetylases antagonized FGF2 and IL-1beta effects on MMP expression in human articular chondrocytesGrowth Factors2009271404919107653
- BradleyKHKawanamiOFerransVJCrystalRGThe fibroblast of human lung alveolar structures: a differentiated cell with a major role in lung structure and functionMethods Cell Biol198021A37646997687
- ZhuYKLiuXDSkoldMCCytokine inhibition of fibroblast-induced gel contraction is mediated by PGE(2) and NO acting through separate parallel pathwaysAm J Respir Cell Mol Biol200125224525311509336
- SatoTLiuXNelsonAReduced miR-146a increases prostaglandin E(2)in chronic obstructive pulmonary disease fibroblastsAm J Respir Crit Care Med201018281020102920522791
- FredrikssonKLundahlJPalmbergLRed blood cells stimulate human lung fibroblasts to secrete interleukin-8Inflammation2003272717812797546