
Abstract
Almost 47 million people suffer from dementia worldwide, with an estimated new case diagnosed every 3.2 seconds. Alzheimer’s disease (AD) accounts for approximately 60%–80% of all dementia cases. Given this evidence, it is clear dementia represents one of the greatest global public health challenges. Currently used drugs alleviate the symptoms of AD but do not treat the underlying causes of dementia. Hence, a worldwide quest is under way to find new treatments to stop, slow, or even prevent AD. Besides the classic targets of the oldest therapies, represented by cholinergic and glutamatergic systems, β-amyloid (Aβ) plaques, and tau tangles, new therapeutic approaches have other targets. One of the newest and most promising strategies is the control of reactive gliosis, a multicellular response to brain injury. This phenomenon occurs as a consequence of a persistent glial activation, which leads to cellular dysfunctions and neuroinflammation. Reactive gliosis is now considered a key abnormality in the AD brain. It has been demonstrated that reactive astrocytes surround both Aβ plaques and tau tangles. In this condition, glial cells lose some of their homeostatic functions and acquire a proinflammatory phenotype amplifying neuronal damage. So, molecules that are able to restore their physiological functions and control the neuroinflammatory process offer new therapeutic opportunities for this devastating disease. In this review, we describe the role of neuroinflammation in the AD pathogenesis and progression and then provide an overview of the recent research with the aim of developing new therapies to treat this disorder.
Introduction
Dementia is a chronic condition characterized by a progressive cognitive impairment that leads to functional disability.Citation1 In 2015, it was estimated that approximately 47 million people worldwide were affected by dementia, and this number is expected to increase, reaching 131.5 million by 2050.Citation2 As such, it represents a veritable public health challenge. Alzheimer’s disease (AD), a pathology first described by Alois Alzheimer in 1907,Citation3 is the most frequent cause of dementia in elderly. Knowledge about the etiology and pathogenesis of the disease is continuously updated,Citation4 but there are still limitations in diagnostic capabilityCitation5 and in the discovery of pharmacological treatments that would be able to stop or better prevent the disease. At present, AD is incurable. Despite the huge amount of preclinical and clinical investigation, medications currently used provide only a modest symptomatic relief to a subset of patients and do not treat the underlying causes of this disease. The reasons for this failure are probably due to the scant knowledge of the cellular and molecular mechanisms implicated in AD pathogenesis and of the approved therapies that coarsely affect both cholinergic and glutamatergic neurotransmission. Conversely, many of the new drugs in development aim to modify the disease process itself by impacting one or more of the many wide-ranging brain changes caused by AD. These changes offer potential targets for new drugs to stop or slow down the disease progression. It is now well recognized that AD is a multifactorial disorder. It is pathologically characterized by widespread oxidative stress, mitochondrial damage, glutamate excitotoxicity, neuroinflammation, neurofibrillary tangle (NFT) formation, and β-amyloid (Aβ) deposition creating senile plaques (SPs).Citation6 These latter are constituted by Aβ peptide, and their genesis is followed by intracellular deposition of NFTs,Citation7 as a consequence of tau protein hyperphosphorylation. The results are synaptic and neuronal dysfunction and loss.Citation8 Over the years, it has been demonstrated that other factors play an important role in the pathogenesis and progression of AD. Among them, the key role of neuroinflammation has been affirmed.Citation9
Physiologically, the inflammatory process is aimed at controlling injuries through several mechanisms to repair tissues.Citation10 However, an increasing amount of literature confirms its role in the pathogenesis and exacerbation of AD.Citation11–Citation14 Inflammation acts to remove both the initial cause of the infliction and to eliminate the destroyed tissues and dead cells resulting from the original injury.
In fact, inflammation is emerging as the real cause of the associated disease, more than a mere contribution to the exacerbation of tissue damage. Indeed, some studies have revealed that the injection of lipopolysaccharide in transgenic mice induces neuroinflammation, triggering intracellular Aβ deposit and tau phosphorylation.Citation15,Citation16
The molecular processes are not necessarily the primary events. The inflammatory machine could also be triggered by traumatic or surgical causes. The microglial priming model suggests that the presymptomatic AD pathology, characterized by low levels of proinflammatory mediators, can act on microglia for long periods of time.Citation17 Furthermore, stress, inflammation, and infection can operate as secondary triggers, causing changes in these primed cells: they reach an activated state establishing an inflammatory response contributing to AD pathogenesis.Citation18
From an immunological point of view, the central nervous system was always seen as a highly protected tissue, exposed to inflammatory phenomena solely in cases of infection or disruption of the blood–brain barrier (BBB). Nowadays, we know that there are several cells expressing pattern recognition receptors able to induce inflammatory signaling pathways.Citation13 These pattern recognition receptors can recognize molecular signals of microbial molecules, called pathogen-associated molecular patterns, as well as endogenous damage-associated molecular patterns (DAMPs), that typically accumulate in infected tissues. DAMPS are present in diseased brains as misfolded proteins (eg, SPs and NFTs), aggregated peptides, or nucleic acids.Citation19 It is clear that DAMPs can trigger neuroinflammation by deflecting proinflammatory reactions from their helpful purpose, and this is the reason why our way of viewing neurodegenerative diseases has changed over the years.
The role of the neuroinflammatory process is not exclusively attributable to innate immunity (which in the brain is constituted by microglia), but it is also caused by other brain resident cells that constitute, in one word, macroglia (ie, astrocytes, NG2-positive cells, and oligodendrocytes), as well as endothelial cells and neurons.Citation20–Citation23
Hence, it is clear that there are many characters involved in this inflammatory process. Thus, a better knowledge of the mechanisms underlying the role of neuroinflammation in AD can be an excellent starting point for the development of molecules able to counteract it.
The pathophysiology of neuroinflammation and its role in Alzheimer’s disease
Even if Aβ deposits can alone induce an inflammatory response that subsequently leads to AD development, it is well established that the neuroinflammatory pathophysiology is more complex and driven by the activation of different brain cells. In particular, growing evidence suggests that this phenomenon is mainly supported by glial cells, which respond quickly to brain injuries, activating a series of repair mechanisms to restore brain physiology. Glial cells are nonexcitable cells of the central nervous system. These cells are a highly heterogeneous population, responsible for many important brain functions.Citation24 While microglia acts as the first form of immune defense in the brain, astrocytes are an essential neurosupportive cell type. Indeed, astrocytes finely control the environment by regulating pH, ion homeostasis, oxidative stress, and blood flow.Citation25,Citation26 These cells together with microglia, oligodendrocytes, neurons, pericytes, and endothelial cells constitute the neurovascular unit, responsible for the proper functioning of the BBB.Citation27 In addition, astrocytes contribute importantly to synaptogenesis and dynamically modulate information processing and signal transmission, regulate neural and synaptic plasticity, and provide trophic and metabolic support to neurons.Citation28,Citation29 Interestingly, data from animal models and human autopsy revealed that both SPs and NFTs cause an immune response in the brain and colocalize close to activated glial cells. Astrocyte and microglia acquire a reactive phenotypeCitation20 and rapidly act in response to pathology undergoing important changes in their morphology and functioning.Citation30,Citation31 Such an activation is fundamentally a protective response aimed at removing injurious stimuli. The neuroprotective action of reactive astrocytes takes place by modulating Aβ-mediated neurotoxicity, degrading, internalizing, and removing Aβ, thus creating a protective barrier that surrounds plaques.Citation32–Citation34 However, uncontrolled and prolonged activation goes beyond physiological control, and detrimental effects override the beneficial ones. In this condition, glial cells foster neuroinflammatory response, accounting for the synthesis of different cytokines and proinflammatory mediators.Citation35,Citation36 This condition is called reactive gliosis and is a characteristic event of AD brains (). For example, activated microglia reduces Aβ accumulation by increasing its phagocytosis, clearance, and degradation,Citation37 as well as by secreting factors such as the glia-derived neurotrophic factor, helpful for neuronal survival.Citation38 Recently, microglia functions aimed at Aβ clearance were attributed to the presence of triggering receptor expressed on myeloid cells 2 (TREM2), a transmembrane receptor;Citation39,Citation40 indeed not long ago, TREM2 was identified as a risk gene for AD.Citation41,Citation42 It is reasonable to think that this association between TREM2 and AD is due to the many functions carried out through the activation of different pathways ranging from phagocytosis to encouraging survival and proliferation, and finally promoting secretion of cytokines and chemokines.Citation43–Citation45
Figure 1 Schematic representation of glial activation.
Notes: As a result of brain damage (eg, brain trauma, ischemia, Aβ accumulation, NFTs, etc) microglia and astrocytes acquire a so-called reactive phenotype losing their physiological functions. Morphofunctional changes, loss of three-dimensional network, and neurovascular unit alterations contribute to cause a homeostatic imbalance. Moreover, after activation, these cells produce a wide range of cytokines and proinflammatory mediators, leading to chronic inflammation. Even if the initial intent of these modifications is reparative, such long-lasting and uncontrolled activation causes further neurodegeneration.
Abbreviations: ROS, reactive oxygen species, NO, nitric oxide; Aβ, β-.amyloid; NFTs, neurofibrillary tangles; BBB, blood–brain barrier.
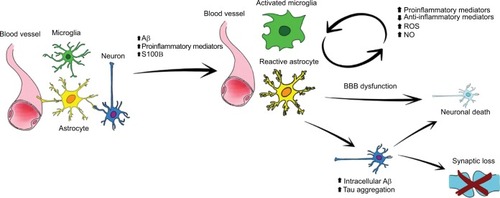
Even astrocytes play an important role in the maintenance of the cerebral homeostasis. These cells are responsible for the proper functioning of the BBB, provide nutrients to neurons, preserve the extracellular ion balance, and remove and degrade Aβ.Citation46 However, glial functions are deeply altered whenever tissue physiology is not restored. In these circumstances, the inability to counteract Aβ and NFTs accumulation constantly stimulates the machinery needed to remove debris; in this way, astrocytes actively support inflammation.Citation47,Citation48
Several studies demonstrated that their action becomes relevant from early stages of the pathogenic process, turning to a cycle independent from Aβ presence, neural dysfunction, cell death, and disease progression.Citation49–Citation51 The resulting chronic inflammation is due to the release of proinflammatory molecules that act not only in an autocrine manner, allowing the perpetuation of the reactive gliosis, but also in a paracrine one, the main cause of the neuronal death that increases the pathological damage.Citation52,Citation53 Neuronal death is determined by the release of not only inflammatory mediators, but also of reactive oxygen species, nitric oxide (NO), proteolytic enzymes, complement factors, and/or excitatory aminoacids.Citation54 At the molecular level, the release of these mediators affects neuron–glia crosstalk, influencing redox enzyme sensors, receptors, and transcription factors.Citation55
In physiological conditions, microglia protects the brain from pathogens, and, together with macroglia, helps maintain homeostasis of the tissue. In AD, all these cells became more reactive and change their morphology surrounding SPs.Citation56 This is possible because of the presence of proinflammatory receptors on their surface. Microglia is able to identify and bind Aβ oligomers and fibrils and the amyloid precursor protein (APP)Citation57 through a large number of receptors, including scavenger receptor class A type 1, MARCO, scavenger receptor class B member 1, CD36, and the receptor for advanced glycation end product,Citation58,Citation59 G protein-coupled receptors formyl peptide receptor 2 Citation60 and chemokine-like receptor 1,Citation61 toll-like receptors (TLRs) TLR2,Citation62 TLR4, and the CD14 coreceptor, and α6β1 integrin.Citation63 The outcome of the bond between Aβ and these receptors is the production of inflammatory mediators such as cytokines (interleukin [IL]-1α, IL-1β, IL-6, IL-8, IL-12, IL-18, and IL-23, interferon (IFN)-γ, tumor necrosis factor [TNF]-α, and granulocyte-macrophage colony-stimulating factor [GM-CSF]),Citation64,Citation65 chemokines (monocyte chemotactic protein 1 (MCP1), MCP-113, fractalkine),Citation66,Citation67 chemoattractant proteins, prostaglandins, complement factors, thromboxanes, pentraxins, NO, reactive oxygen species, leukotrienes, proteases, protease inhibitors, adhesion molecules (interaction between CD40-CD40 ligand CD40L),Citation68 coagulation factors, and C-reactive protein, most of which are detectable in AD animal and/or in the brain or cerebrospinal fluid of AD patients.Citation25,Citation69,Citation70 However, glial cells are also capable of producing some regulatory cytokines, such as IL-10 and transforming growth factor-β (TGF-β), but in AD their release is modified, exacerbating the disease.Citation71–Citation73 Among anti-inflammatory factors, we also recall the cluster of differentiation-200 (CD200) regulated by the anti-inflammatory IL-4 and expressed by neurons, T- and B-cells, whose receptor is expressed by glia. Both AD patients and mouse models show an age-related or Aβ-induced CD200 reduction.Citation74–Citation76
Upstream of cytokines production is the activation of the nuclear factor-kappa B (NF-κB) pathway,Citation77 and the subsequent activation of mitogen-activated protein kinase (MAPK) pathways, whose proinflammatory gene expression is Aβ dependent.Citation78 Extracellular signal-regulated protein kinases (ERKs), stress-activated protein kinases c-Jun NH2-terminal kinase (JNK), and p38 constitute the set of MAPKs whose action is exerted both in the cytoplasm and in the nucleus, thereby phosphorylating transcription factors. For example, p38 can contribute to neuroinflammation by inducing TNF-α gene transcription, which increases the activator protein-1 (AP-1) activity,Citation79 besides being directly responsible for tau phosphorylation.Citation80
In turn, proinflammatory mediators increase the activity and the products of amyloidogenic pathway, especially Aβ(1–42). For instance, the γ-secretase cell-based assays showed that TNF-α, IL-1β, and IFN-γ cause the initiation of APP cleavage through the MAPK pathway,Citation81 and a more recent study demonstrated that NF-κB signaling, activated by TNF-α, results in an increased Aβ synthesis driven by the β-secretase (BACE-1) transcription.Citation82
To the vicious circle driven by cytokines and MAPKs,Citation83,Citation84 the resulting activation of the complement cascade has to be added,Citation85 as well as the induction of proinflammatory enzymes, such as cyclooxygenase-2 (COX-2)Citation86 and the inducible nitric oxide synthase (iNOS).Citation87 Induction of these enzymes may also be linked to the excessive release of S100B (ββ form of the S100), a neurotrophin expressed by activated astrocytes,Citation88 which is able to induce NF-κB activation,Citation89 as well as encourage tauopathy.Citation23 Two more proinflammatory proteins, implicated in the pathophysiology of AD, belong to the S100 family: S100A9 and S100A12. These proteins, produced by activated microglia and macrophages, are increased in AD brain and are responsible for protein complex formation.Citation90,Citation91 S100A9 is present within SPs and Aβ deposits surrounding blood vessels, and it is also abundant in tissues neighboring Aβ deposits, confirming that increased S100A9 levels can stimulate peptides aggregation and deposition.Citation92
Studies report that tau hyperphosphorylation is directly affected by inflammatory mediators, including the cyclin-dependent kinase 5 (CDK5):Citation93 IL-6 stimulates neuronal protein p35, which in turn is responsible for the kinase activation that can act on tau.Citation94 CDK5 is not the only kinase related to neuroinflammation. Recently, the role of protein kinase 2 (CK2, former casein kinase II) has been described. In fact, CK2 immunopositive astrocytes have been found to be associated with amyloid deposits in AD brains, suggesting its involvement in the neuroinflammatory response.Citation95
Inflammatory mediators, in particular cytokines, are also responsible for increased BBB permeabilization driven by chemokines, allowing leukocyte penetration in the brain.Citation96,Citation97 This is possible because of altering the resistance of tight junctions, upregulation of cytokines expression, and COX-2 transcription in endothelial cells.Citation98 For example, IL-6, IL-10, IL-13, and prostaglandins stimulated by lipopolysaccharide may increase the influx of Aβ across the BBB, besides upregulating APP processing in the brain.Citation99,Citation100
Another mechanism underlying the pathogenic process led by neuroinflammation is the blockage of neurogenesis, which is inhibited by some proinflammatory cytokines such as IL-6, TNF-α, and IL-18, responsible for neural progenitor cells death, and inhibition of their differentiation.Citation101 Interestingly, these cells are located in the subgranular layer of the dentate gyrus of the hippocampus, in the subventricular zone of the lateral ventricles, and amygdala – areas mainly affected by AD and cognitive impairment.Citation102
One of the still poorly explored mechanisms that might govern the relationship between AD and neuroinflammation, but definitely is in charge of the neurodegenerative processes, involves the glycogen synthase kinase-3 (constitutively active serine/threonine protein kinase) pathway.Citation103 This idea comes from the observation of the results obtained by blocking this kinase, which causes an increase of the anti-inflammatory IL-10 and a decrease of proinflammatory cytokines as a consequence of TLRs stimulation and NO production.Citation104–Citation106
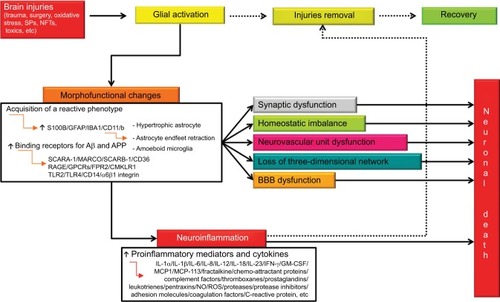
The salient events reported in this paragraph are summarized in .
Figure 2 Integrated pathways between glial activation, neuroinflammation, and neuronal death after brain injury.
Notes: Whenever a brain injury occurs, glial activation takes place with the aim of removing injurious stimuli. To this aim, activated cells undergo a series of morphofunctional changes and acquire a reactive phenotype. Activation causes, among other things, glial hypertrophy, astrocyte endfeet retraction, and gain of amoeboid microglial structure. These changes, if not stopped, can induce synaptic dysfunction, homeostatic imbalance, neurovascular unit dysfunction, loss of three-dimensional network, and BBB dysfunction. In addition, reactive microglia and astrocytes release a wide range of proinflammatory mediators aimed at removing the primary injury. The occurrence of a reactive state is very probably a protective response. However, uncontrolled and prolonged activation goes beyond physiological control, and detrimental effects override the beneficial ones. Solid arrows indicate complete pathways. Dashed arrows indicate pathways that that occur partially or do not.
Abbreviations: BBB, blood–brain barrier; Aβ, β-amyloid; NFTs, neurofibrillary tangles; SCARA-1, scavenger receptor class A Type 1; SCARB-1, scavenger receptor class B member 1; RAGE, receptor for advanced glycation end product; GPCRs, G protein-coupled receptors; FPR2, formyl peptide receptor 2; CMKLR1, chemokine-like receptor 1; TLRs, toll-like receptors; IL, interleukin; IFN-γ, interferon-γ; GM-CSF, granulocyte-macrophage colony-stimulating factor; NO, nitric oxide; ROS, reactive oxygen species; SPs, senile plaques; IBA1, ionized calcium-binding adapter molecule 1; CD, cluster of differentiation.
Neuroinflammatory targets in Alzheimer’s disease
Because of the knowledge acquired so far and the failure of so many antiamyloid trials, scientific interest has shifted to other features of neurodegeneration including neuroinflammation.Citation107
Evidence mentioned in “The Pathophysiology of Neuroinflammation and Its Role in Alzheimer’s Disease” section shows how neuroinflammation is driven by a large number of events apparently different but strongly dependent one on the other.Citation108 For this reason, it is difficult to identify the best target upon which to act. Recently, much work has been done, but much more research still needs to be done.
It is now clear that the AD neurodegenerative process is also orchestrated by proinflammatory cytokines and their receptors, which therefore become promising targets on which to focus by means of different approaches. Blocking gene expression of cytokines, releasing or binding their receptors, or better regulating the functioning of cells implicated in the neuroinflammation are definitely strategies still in exploration.Citation109 The possibility of of reducing tau kinase activity and oligomeric and fibrillary Aβ accumulation by neutralizing IL-1β or TNF-α/TNF-α receptor through antibodies has been demonstrated in murine models of AD.Citation64,Citation110–Citation113 In this context, the role of molecules with anti-inflammatory properties (such as minocycline) that are able to decrease astrocyte release of proinflammatory cytokines and reduce both tau and amyloid pathogenesis,Citation114 as well as improve AD behavioral symptoms, is not less important.Citation115
Interestingly, both in vitro and in vivo studies have shown that pharmacological inhibition of COX-2 and inducible NO synthase has positive outcomes.Citation38,Citation116–Citation120
Lastly, in AD models it was observed that it is possible to obtain satisfactory results by modulating kinases that are not only directly related to tau hyperphosphorylation but also to neuroinflammation. One example is the modulation of glycogen synthase kinase-3β. Experimental studies have shown that it is possible to exert anti-inflammatory effect by inhibiting this enzyme, giving us another potential therapeutic target to consider.Citation87,Citation88,Citation121
Currently available products and products in research and development focusing on neuroinflammatory targets
In the past decades, several epidemiological and clinical studies were carried out to demonstrate the neuroprotective potential of several nonsteroidal anti-inflammatory drugs.Citation122,Citation123 After the pioneering work with indometacin demonstrated the ability to restore cognitive functions in the enrolled subjects, many other clinical trials have shown only unsatisfactory results.Citation124–Citation127 Since the failure of trials with classical nonsteroidal anti-inflammatory drugs, scientists tested COX-2-selective compounds effects. Once again, results were disappointing.Citation128,Citation129 Evidence from a clinical trial with naproxen suggests its ability to reduce tau and Aβ levels in cerebrospinal fluid and plasma.Citation130 AD is a multifactorial disease and the inflammatory outcome, driven by glial activation, depends on the context and on the stage of the pathology. For these reasons, an ideal anti-inflammatory compound should be able to control the detrimental effects and, at the same time, preserve the physiological glial activation.
An alternative and recent therapeutic approach is represented by nutraceuticals (eg, curcumin, apigenin, docosahexaenoic acid, resveratrol, and n-3 fatty acids).Citation131–Citation134 Despite encouraging preclinical results, the success rate in humans has been very low.Citation135
Complex results were obtained after vaccinating AD patients against Aβ and NFTs. A large number of studies have been done in this field, and promising data were obtained in preclinical models. Unfortunately, these encouraging findings were not replicated in clinical trials, and promising vaccines were stopped because of adverse effects such as meningoencephalitis.Citation136 Some of these studies revealed that immunization halts glial activation.Citation137 By the physiological importance of this phenomenon, this is probably why the immunization has caused severe adverse reactions.
Presently, several competing hypotheses (especially related to time of intervention) may help explain the failure of translating preclinical studies into the clinical ones, but so far there is no way to confirm which of these explanations is correct.
Future research direction
The pathogenic role of neuroinflammation in AD is now well recognized and accepted. Nevertheless, the underlying mechanisms have not been sufficiently elucidated. Several factors contribute to this failure. First of all, there is a lack of adequate preclinical models that best mimic the disease and, in particular, the processes of glial activation and neuroinflammation. Then, another important factor is the comprehension of the role of each cellular component in the inflammatory process, for example, the identification of cell-specific biomarkers. Indeed, specifically clarifying changes in both immune system and inflammatory machinery would make available different pathways for pharmacological manipulations aimed at delaying the onset and/or the progression of the disease. Finally, it is important to define the inflammatory stages to correlate each phase to AD progression and to clarify which processes are protective and which ones are detrimental.
The achievement of these goals will allow scientists to practice many other experimental approaches. The hope is to get satisfactory results from clinical studies with compounds that have been successful in vitro, ex vivo, and/or in vivo experiments, such as the administration of molecules like acetylpuerarin,Citation138 edaravone,Citation139 palmitoylethanolamide,Citation38 N-[2-(4-hydroxyphenyl)ethyl]-2-(2,5-dimethoxyphenyl)-3-(3-methoxy-4-hydroxyphenyl) acrylamide (compound FLZ),Citation140 oleuropeinaglycone,Citation141 oridonin,Citation142 protocatechuic acid,Citation143 resveratrol,Citation110 rutin,Citation144 or immunotherapiesCitation145,Citation146 and vaccinations.Citation147
Conclusion
Growing evidence confirms that neuroinflammation, finely orchestrated by neuronal, glial, and immune components, is a contributing cause of Aβ aggregation, tau hyperphosphorylation, and neuronal damage and death. The resulting production of cytokines and proinflammatory molecules has initially a neuroprotective role, but subsequently becomes the cause of further neurodegeneration.
Unfortunately, because of the lack of appropriate animal models, we still lack a complete understanding of the relationship between inflammatory process stages and AD progression. This could explain, at least in part, the unsuccessful results of clinical trials performed with anti-inflammatory molecules whose efficacy was significantly proven in preclinical investigations.
Therefore, future experimental studies must intensively investigate the intricate paths of the neuroinflammatory process and define the best time to control it. In this way, it will be possible to achieve more focused and functional therapeutic strategies in the hope of not only alleviating but also modifying AD progression.
Acknowledgments
This work was supported by the SAPIENZA University Grant to CS (prot. C26A15X58E). The authors thank Miss Federica Bellifemine for her helpful contribution in preparing the .
Disclosure
The authors report no conflicts of interest in this work.
References
- StefaniakJO’BrienJImaging of neuroinflammation in dementia: a reviewJ Neurol Neurosurg Psychiatry2016871212826384512
- PrinceMWimoAGuerchetMAliGCWuYTPrinaMWorld Alzheimer Report 2015. The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and TrendsAlzheimer’s Disease International2015
- AlzheimerAUber eine eigenartige Erkangkung der Hirnrinde (An unusual illness of the cerebral cortex)Allgemeine Zeitschr Psychisch-Gerichtliche Medizin190764146148
- SperlingRAAisenPSBeckettLAToward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s diseaseAlzheimers Dement2011728029221514248
- LopezOLSchwamECummingsJPredicting cognitive decline in Alzheimer’s disease: an integrated analysisAlzheimers Dement2010643143921044773
- QuerfurthHWLaFerlaFMAlzheimer’s diseaseN Engl J Med2010362432934420107219
- BraakHBraakEMorphological criteria for the recognition of Alzheimer’s disease and the distribution pattern of cortical changes related to this disorderNeurobiol Aging1994355356 discussion 379–3807936061
- BlennowKde LeonMJZetterbergHAlzheimer’s diseaseLancet200636838740316876668
- FerreiraSTClarkeJRBomfimTRDe FeliceFGInflammation, defective insulin signaling, and neuronal dysfunction in Alzheimer’s diseaseAlzheimers Dement2014101 SupplS76S8324529528
- BrownKLCosseauCGardyJLHancockREComplexities of targeting innate immunity to treat infectionTrends Immunol20072826026617468048
- ProkopSMillerKRHeppnerFLMicroglia actions in Alzheimer’s diseaseActa Neuropathol201312646147724224195
- PerryVHHolmesCMicroglial priming in neurodegenerative diseaseNature Rev Neurol20141021722424638131
- HenekaMTKummerMPLatzEInnate immune activation in neurodegenerative diseaseNature Rev Immunol20141446347724962261
- ZhangBGaiteriCBodeaLGIntegrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s diseaseCell201315370772023622250
- ShengJGBoraSHXuGBorcheltDRPriceDLKoliatsosVELipopolysaccharide induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic miceNeurobiol Dis20031413314513678674
- KitazawaMOddoSYamasakiTRLipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s diseaseJ Neurosci2005258843885316192374
- VukicVCallaghanDWalkerDExpression of inflammatory genes induced by beta-amyloid peptides in human brain endothelial cells and in Alzheimer’s brain is mediated by the JNK-AP1 signaling pathwayNeurobiol Dis2009349510619162185
- CunninghamCCampionSLunnonKSystemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative diseaseBiol Psychiatry20096530431218801476
- VezzaniMMarosoMBalossoSSanchezMABartfaiTIL-1 receptor/Toll-like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizuresBrain Behav Immun20112571281128921473909
- VerkhratskyARodríguezJJSteardoLAstrogliopathology: a central element of neuropsychiatric diseases?Neuroscientist201420657658824301046
- SteardoLJrBronzuoliMRIacominoAEspositoGSteardoLScuderiCDoes neuroinflammation turn on the flame in Alzheimer’s disease? Focus on astrocytesFront Neurosci2015925926283900
- LécuyerMAKebirHPratAGlial influences on BBB functions and molecular players in immune cell traffickingBiochim Biophys Acta20161862347248226454208
- EspositoGScuderiCLuJS100B induces tau protein hyperphosphorylation via Dickopff-1 up-regulation and disrupts the Wnt pathway in human neural stem cellsJ Cell Mol Med200812391492718494933
- VerkhratskyANedergaardMHertzLWhy are astrocytes important?Neurochem Res201540238940125113122
- DeitmerJWRoseCRpH regulation and proton signalling by glial cellsProg Neurobiol1996482731038737439
- IadecolaCNedergaardMGlial regulation of the cerebral microvasculatureNat Neurosci200710111369137617965657
- ZlokovicBVThe blood-brain barrier in health and chronic neurodegenerative disordersNeuron200857217820118215617
- PereaGNavarreteMAraqueATripartite synapses: astrocytes process and control synaptic informationTrends Neurosci200932842143119615761
- SofroniewMVAstrogliosis: Diversity of astrocyte functions and phenotypes in neural circuitsCold Spring Harb Perspect Biol201472a02042025380660
- SofroniewMVVintersHVAstrocytes: biology and pathologyActa Neuropathol2010119173520012068
- ScuderiCSteccaCIacominoASteardoLRole of astrocytes in major neurological disorders: the evidence and implicationsIUBMB Life2013651295796124376207
- ParadisiSSacchettiBBalduzziMGaudiSMalchiodi-AlbediFAstrocyte modulation of in vitro beta-amyloid neurotoxicityGlia200446325226015048848
- ThalDRThe role of astrocytes in amyloid beta-protein toxicity and clearanceExp Neurol20122361522575598
- MathurRIncePGMinettTA reduced astrocyte response to β-amyloid plaques in the ageing brain associates with cognitive impairmentPLoS One2015102e011846325707004
- MrakREGriffinWSThe role of activated astrocytes and of the neurotrophic cytokine S100B in the pathogenesis of Alzheimer’s diseaseNeurobiol Aging200122691592211754999
- TuppoEEAriasHRThe role of inflammation in Alzheimer’s diseaseInt J Biochem Cell Biol200537228930515474976
- FrautschySAYangFIrrizarryMMicroglial response to amyloid plaques in APPsw transgenic miceAm J Pathol19981523073179422548
- LiuBHongJSRole of Microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic interventionJ Pharm Exp Ther200330417
- MelchiorBGarciaAEHsiungBKDual induction of TREM2 and tolerance-related transcript, Tmem176b, in amyloid transgenic mice: implications for vaccine-based therapies for Alzheimer’s diseaseASN Neuro201023e0003720640189
- JiangTTanLZhuXCUpregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer’s diseaseNeuropsychopharmacol2014391329492962
- GuerreiroRWojtasABrasJTREM2 variants in Alzheimer’s diseaseN Engl J Med201336811712723150934
- JonssonTStefanssonHSteinbergSVariant of TREM2 associated with the risk of Alzheimer’s diseaseN Engl J Med201336810711623150908
- TakahashiKRochfordCDNeumannHClearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2J Exp Med200520164765715728241
- OteroKShinoharaMZhaoHTREM2 and β-catenin regulate bone homeostasis by controlling the rate of osteoclastogenesisJ Immunol201218862612262122312126
- BouchonAHernandez-MunainCCellaMColonnaMADAP12-mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cellsJ Exp Med20011941111112211602640
- Wyss-CorayTLoikeJDBrionneTCAdult mouse astrocytes degrade amyloid-beta in vitro and in situNat Med2003945345712612547
- Rubio-PerezJMMorillas-RuizJMA review: inflammatory process in Alzheimer’s disease, role of cytokinesScientific World J20122012756357
- TownTNikolicVTanJThe microglial “activation” continuum: from innate to adaptive responsesJ Neuroinflammation200522416259628
- GandySHeppnerFLMicroglia as dynamic and essential components of the amyloid hypothesisNeuron20137857557723719156
- SudduthTLSchmittFANelsonPTWilcockDMNeuroinflammatory phenotype in early Alzheimer’s diseaseNeurobiol Aging2013341051105923062700
- HolmesCCunninghamCZotovaESystemic inflammation and disease progression in Alzheimer diseaseNeurology20097376877419738171
- ScuderiCSteccaCValenzaMPalmitoylethanolamide controls reactive gliosis and exerts neuroprotective functions in a rat model of Alzheimer’s diseaseCell Death Dis20145e141925210802
- BlockMLHongJSMicroglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanismProg Neurobiol200576779816081203
- ChristovAOttmanJTGrammasPVascular inflammatory, oxidative and protease-based processes: implications for neuronal cell death in Alzheimer’s diseaseNeurol Res200426554054615265271
- LiuXWuZHayashiYNakanishiHAge-dependent neuroinflammatory responses and deficits in long-term potentiation in the hippocampus during systemic inflammationNeuroscience201221613314222554776
- OlabarriaMNoristaniHNVerkhratskyARodríguezJJConcomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s diseaseGlia201058783183820140958
- BargerSWHarmonADMicroglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein ENature199738866458788819278049
- YuYYeRDMicroglial Aβ receptors in Alzheimer’s diseaseCell Mol Neurobiol2015351718325149075
- CaiZLiuNWangCRole of RAGE in Alzheimer’s DiseaseCell Mol Neurobiol201636448349526175217
- LeYGongWTiffanyHLAmyloid (beta)42 activates a G-protein-coupled chemoattractant receptor, FPR-like-1J Neurosci2001212RC12311160457
- PengLYuYLiuJThe chemerin receptor CMKLR1 is a functional receptor for amyloid-beta peptideJ Alzheimers Dis2014431227242
- StewartCRStuartLMWilkinsonKCD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimerNature Immunol20101115516120037584
- KoenigsknechtJLandrethGMicroglial phagocytosis of fibrillar β-amyloid through a β1 integrin-dependent mechanismJ Neurosci2004498389846
- von BernhardiRTichauerJEEugenínJAging dependent changes of microglial cells and their relevance for neurodegenerative disordersJ Neurochem20101121099111420002526
- LiKLiuSYaoSWangBDaiDYaoLInteraction between interleukin-8 and methylenetetrahydrofolate reductase genes modulates Alzheimer’s disease riskDement Geriatr Cogn Disord20092728629119246914
- WestinKBuchhavePNielsenHMinthonLJanciauskieneSHanssonOCCL2 is associated with a faster rate of cognitive decline during early stages of Alzheimer’s diseasePLoS One20127e3052522303443
- WuJBieBYangHXuJJBrownDLNaguibMSuppression of central chemokine fractalkine receptor signaling alleviates amyloid-induced memory deficiencyNeurobiol Aging2013342843285223855980
- YuSLiuYPLiuYHDiagnostic utility of VEGF and soluble CD40L levels in serum of Alzheimer’s patientsYu Clin Chim Acta201645315415926706786
- AlamQAlamMZMushtaqGInflammatory process in Alzheimer and Parkinson’s diseases: central role of cytokinesCurr Pharm Des201622554154826601965
- ShenYMeriSYin and Yang: complement activation and regulation in Alzheimer’s diseaseProg Neurobiol200370646347214568360
- WangWYTanMSYuJTTanLRole of pro-inflammatory cytokines released from microglia in Alzheimer’s diseaseAnn Transl Med201531013626207229
- SierraAGottfried-BlackmoreACMcewenBSBullochKMicroglia derived from aging mice exhibit an altered inflammatory profileGlia20075541242417203473
- Welser-AlvesJVMilnerRMicroglia are the major source of TNF-alpha and TGF-beta1 in post natal glial cultures; regulation by cytokines, lipopolysaccharide and vitronectinNeurochem Int201363475323619393
- VarnumMMKiyotaTIngrahamKLIkezuSIkezuTThe anti-inflammatory glycoprotein, CD200, restores neurogenesis and enhances amyloid phagocytosis in a mouse model of Alzheimer’s diseaseNeurobiol Aging201536112995300726315370
- LyonsAMcQuillanKDeighanBDecreased neuronal CD200 expression in IL-4-deficient mice results in increased neuroinflammation in response to lipopolysaccharideBrain Behav Immunol200923710201027
- WalkerDGDalsing-HernandezJECampbellNALueLFDecreased expression of CD200 and CD200 receptor in Alzheimer’s disease: a potential mechanism leading to chronic inflammationExp Neurol2009215151918938162
- WuDZhangXZhaoMZhouALThe role of the TLR4/NF-κB signaling pathway in Aβ accumulation in primary hippocampal neuronsActa Physiologica Sinica201567331932826109305
- FeldMKrawczykMCSol FustiñanaMDecrease of ERK/MAPK overactivation in prefrontal cortex reverses early memory deficit in a mouse model of Alzheimer’s diseaseJ Alzheimers Dis2014401698224334722
- SpriggsDRDeutschSKufeDWGenomic structure, induction and production of TNF-alphaImmunol Ser1992563341550865
- FeijooCCampbellDGJakesRGoedertMCuendaAEvidence that phosphorylation of the microtubule-associated protein Tau by SAPK4/p38delta at Thr50 promotes microtubule assemblyJ Cell Sci200511839740815632108
- LiaoYFWangBJChengHTKuoLHWolfeMSTumor necrosis factor-alpha, interleukin-1beta, and interferon-gamma stimulate gamma-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathwayJ Biol Chem20042794749523493215347683
- ChenCHZhouWLiuSIncreased NF-kappaB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s diseaseInt J Neuropsychopharmacol2012151779021329555
- MunozLAmmitAJTargeting p38 MAPK pathway for the treatment of Alzheimer’s diseaseNeuropharmacology201058356156819951717
- GlassCKSaijoKWinnerBMarchettoMCGageFHMechanisms underlying inflammation in neurodegenerationCell2010140691893420303880
- LianHLitvinchukAChiangACAithmittiNJankowskyJLZhengHAstrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer’s diseaseJ Neurosci201636257758926758846
- FattahiMJMirshafieyAPositive and negative effects of prostaglandins in Alzheimer’s diseasePsychiatry Clin Neurosci2014681506023992456
- MalinskiTNitric oxide and nitroxidative stress in Alzheimer’s diseaseJ Alzheimers Dis20071120721817522445
- HuJFerreiraAVan EldikLJS100β induces neuronal cell death through nitric oxide release from astrocytesJ Neurochem199769229423019375660
- MoriT1KoyamaNArendashGWHorikoshi-SakurabaYTanJTownTOverexpression of human S100B exacerbates cerebral amyloidosis and gliosis in the Tg2576 mouse model of Alzheimer’s diseaseGlia201058330031419705461
- ShepherdCEGoyetteJUtterVInflammatory S100A9 and S100A12 proteins in Alzheimer’s diseaseNeurobiol Aging200627111554156316253391
- VoglTGharibyanALMorozova-RocheLAProinflammatory S100A8 and S100A9 proteins: self-assembly into multifunctional native and amyloid complexesInt J Mol Sci20121332893291722489132
- WangCKlechikovAGGharibyanALThe role of pro-inflammatory S100A9 in Alzheimer’s disease amyloid-neuroinflammatory cascadeActa Neuropathol2014127450752224240735
- CzapskiGAGąssowskaMWilkaniecAChalimoniukMStrosznajderJBAdamczykAThe mechanisms regulating cyclin-dependent kinase 5 in hippocampus during systemic inflammatory response: the effect on inflammatory gene expressionNeurochem Int20169310311226806339
- QuintanillaRAOrellanaDGonzalez-BillaultCMaccioniRBInterleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathwayExp Cell Res2004295124525715051507
- RosenbergerAFMorremaTHGerritsenWHIncreased occurrence of protein kinase CK2 in astrocytes in Alzheimer’s disease pathologyJ Neuroinflammation2016131426732432
- TerrandoNErikssonLIRyuJKResolving postoperative neuroinflammation and cognitive declineAnn Neurol201170698699522190370
- EngelhardtBT cell migration into the central nervous system during health and disease: different molecular keys allow access to different central nervous system compartmentsClin Exp Neuroimmunol201017993
- WonDDorovini-ZisKVincentSRCytokines, nitric oxide, and cGMP modulate the permeability of an in vitro model of the human blood–brain barrierExp Neurol2004190244645515530883
- DeaneRDu YanSSubmamaryanRKRAGE mediates amyloid-beta peptide transport across the blood–brain barrier and accumulation in brainNat Med20039790791312808450
- JaegerLBDohguSSultanaRLipopolysaccharide alters the blood–brain barrier transport of amyloid beta protein: a mechanism for inflammation in the progression of Alzheimer’s diseaseBrain Behav Immun200923450751719486646
- LiuYPLinHITzengSFTumor necrosis factor-alpha and interleukin-18 modulate neuronal cell fate in embryonic neural progenitor cultureBrain Res20051054215215816054598
- BernierPJBedardAVinetJLevesqueMParentANewly generated neurons in the amygdala and adjoining cortex of adult primatesProc Natl Acad Sci USA20029917114641146912177450
- PhielCJWilsonCALeeVMKleinPSGSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptidesNature2003423693843543912761548
- MartinMRehaniKJopeRSMichalekSMToll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3Nat Immunol20056877778416007092
- WangMJHuangHYChenWFChangHFKuoJSGlycogen synthase kinase-3beta inactivation inhibits tumor necrosis factor-alpha production in microglia by modulating nuclear factor κB and MLK3/JNK signaling cascadesJ Neuroinflammation201079921194439
- HuangWCLinYSWangCYGlycogen synthase kinase-3negatively regulates anti-inflammatory interleukin-10 for lipopolysaccharide-induced iNOS/NO biosynthesis and RANTES production in microglial cellsImmunology20091281 SupplS275S286
- CastelloMAJeppsonJDSorianoSMoving beyond anti-amyloid therapy for the prevention and treatment of Alzheimer’s diseaseBMC Neurol20141416925179671
- FrautschySAColeGMWhy pleiotropic interventions are needed for Alzheimer’s diseaseMol Neurobiol20104139240920437209
- von BernhardiRCornejoFParadaGEEugenínJRole of TGFβ signaling in the pathogenesis of Alzheimer’s diseaseFront Cell Neurosci2015942626578886
- KitazawaMChengDTsukamotoMRBlocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease modelJ Immunol20111876539654922095718
- ShiJQShenWChenJAnti-TNF-α reduces amyloid plaques and tau phosphorylation and induces CD11c-positive dendritic-like cell in the APP/PS1 transgenic mouse brainsBrain Res2011136823924720971085
- HePChengXStaufenbielMLong-term treatment of thalidomide ameliorates amyloid-like pathology through inhibition of β-secretase in a mouse model of Alzheimer’s diseasePLoS One201382e5509123405115
- TweedieDFergusonRAFishmanKTumor necrosis factor-α synthesis inhibitor 3,6′-dithiothalidomide attenuates markers of inflammation, Alzheimer pathology and behavioral deficits in animal models of neuroinflammation and Alzheimer’s diseaseJ Neuroinflammation2012910622642825
- GarwoodCJCooperJDHangerDPNobleWAntiinflammatory impact of minocycline in a mouse model of tauopathyFront Psychiatry2010113621423446
- ParachikovaAVasilevkoVCribbsDHLaFerlaFMGreenKNReductions in amyloid-beta-derived neuroinflammation, with minocycline, restore cognition but do not significantly affect tau hyperphosphorylationJ Alzheimers Dis201021252754220555131
- ScuderiCSteccaCBronzuoliMRSirtuin modulators control reactive gliosis in an in vitro model of Alzheimer’s diseaseFront Pharmacol201458924860504
- BiccaMACostaRLoch-NeckelGFigueiredoCPMedeirosRCalixtoJBB2 receptor blockage prevents Aβ-induced cognitive impairment by neuroinflammation inhibitionBehav Brain Res201527848249125446751
- GanPZhangLChenYAnti-inflammatory effects of glaucocalyxin B in microglia cellsJ Pharmacol Sci20151281354626003084
- ScuderiCSteardoLNeuroglial roots of neurodegenerative diseases: therapeutic potential of palmitoylethanolamide in models of Alzheimer’s diseaseCNS NeurolDisord Drug Targets20131216269
- CirilloCCapocciaEIuvoneTS100B inhibitor pentamidine attenuates reactive gliosis and reduces neuronal loss in a mouse model of Alzheimer’s diseaseBiomed Res Int2015201550834226295040
- GreenHFNolanYMGSK-3 mediates the release of IL-1β, TNF-α and IL-10 from cortical gliaNeurochem Int201261566667122796213
- AndersenKLaunerLJOttAHoesAWBretelerMMHofmanADo nonsteroidal anti-inflammatory drugs decrease the risk for Alzheimer’s disease? The Rotterdam studyNeurology1995458144114457644037
- GaspariniLOnginiEWenkGNon-steroidal antiinflammatory drugs (NSAIDs) in Alzheimer’s disease: old and new mechanisms of actionJ Neurochem200491352153615485484
- FeldmanHHDoodyRSKivipeltoMRandomized controlled trial of atorvastatin in mild to moderate Alzheimer disease: LEADeNeurology2010741295696420200346
- LyketsosCGBreitnerJCGreenRCNaproxen and celecoxib do not prevent AD in early results from a randomized controlled trialNeurology2007681800180817460158
- MartinBKSzekelyCBrandtJCognitive function over time in the Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxibArch Neurol20086589690518474729
- de JongDJansenRHoefnagelsWNo effect of one-year treatment with indomethacin on Alzheimer’s disease progression: a randomized controlled trialPLoS One200831e147518213383
- ReinesSABlockGAMorrisJCRofecoxib: no effect on Alzheimer’s disease in a 1-year, randomized, blinded, controlled studyNeurology2004621667114718699
- ThalLJFerrisSHKirbyLRofecoxib Protocol 078 study group. A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairmentNeuropsychopharmacol200530612041215
- BreitnerJCBakerLDMontineTJExtended results of the Alzheimer’s disease anti-inflammatory prevention trialAlzheimers Dement20117440241121784351
- BaumLLamCWCheungSKSix-month randomized, placebo controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer diseaseJ Clin Psychopharmacol200828111011318204357
- ShintoLQuinnJMontineTA randomized placebo-controlled pilot trial of omega-3 fatty acids and alpha lipoic acid in Alzheimer’s diseaseJ Alzheimers Dis201438111112024077434
- FavaAPirritanoDPlastinoMThe effect of lipoic acid therapy on cognitive functioning in patients with Alzheimer’s diseaseJ Neurodegener Dis2013201345425326316990
- WitteAVKertiLMarguliesDSFloelAEffects of resveratrol on memory performance, hippocampal functional connectivity, and glucose metabolism in healthy older adultsJ Neurosci201434237862787024899709
- VenigallaMSonegoSGyengesiESharmanMJMünchGNovel promising therapeutics against chronic neuroinflammation and neurodegeneration in Alzheimer’s diseaseNeurochem Int201595637426529297
- PohankaMVaccination to Alzheimer Disease. Is it a promising tool or a blind way?Curr Med Chem201623141432144127087245
- ZotovaEBharambeVCheaveauMInflammatory components in human Alzheimer’s disease and after active amyloid-β42 immunizationBrain201313692677269623943781
- MengQHLouFLHouWXLiuMGuoHZhangXMAcetylpuerarin reduces inflammation and improves memory function in a rat model of Alzheimer’s disease induced by Abeta1-42Pharmazie2013681190490824380241
- JiaoSSYaoXQLiuYHEdaravone alleviates Alzheimer’s disease-type pathologies and cognitive deficitsProc Natl Acad Sci USA2015112165225523025847999
- WuLYBaoXQPangHYSunHZhangDFLZ attenuates learning and memory deficits via suppressing neuroinflammation induced by LPS in miceJ Asian Nat Prod Res201517330631725621771
- CasamentiFGrossiCRigacciSPantanoDLuccariniIStefaniMOleuropeinaglycone: a possible drug against degenerative conditions. In vivo evidence of its effectiveness against Alzheimer’s diseaseJ Alzheimers Dis201545367968825649656
- WangSYangHYuLOridonin attenuates Aβ1-42-induced neuroinflammation and inhibits NF-κB pathwayPLoS One201498e10474525121593
- SongYCuiTXieNZhangXQianZLiuJProtocatechuic acid improves cognitive deficits and attenuates amyloid deposits, inflammatory response in aged AβPP/PS1 double transgenic miceInt Immunopharmacol201420127628124667368
- XuPXWangSWYuXLRutin improves spatial memory in Alzheimer’s disease transgenic mice by reducing Aβ oligomer level and attenuating oxidative stress and neuroinflammationBehav Brain Res201426417318024512768
- CantarellaGDi BenedettoGPuzzoDNeutralization of TNFSF10 ameliorates functional outcome in a murine model of Alzheimer’s diseaseBrain2015138120321625472798
- CountsSERayBMufsonEJPerezSEHeBLahiriDKIntravenous immunoglobulin (IVIG) treatment exerts antioxidant and neuropreservatory effects in preclinical models of Alzheimer’s diseaseJ Clin Immunol2014341 SupplS80S8524760109
- XingXNZhangWGShaSAmyloid β 3-10 DNA vaccination suggests a potential new treatment for Alzheimer’s disease in BALB/c miceChin Med J (Engl)2011124172636264122040416