
Abstract
Wiskott–Aldrich syndrome (WAS) is a rare X-linked recessive inborn error of immunity (IEI) first described in 1937. Classic WAS is characterized by the triad of thrombocytopenia with small platelets, recurrent infections due to combined immunodeficiency, and eczema. Hematopoietic stem cell transplantation (HSCT) was the only curative option available for five decades, with excellent outcomes reported for matched sibling donors (MSD) and matched unrelated donors (MUD). More recently, alternative donor transplants such as umbilical cord blood (UCB) and haploidentical transplant have emerged as viable options due to improvements in better graft selection, cell dosing, and effective allograft manipulation measures. Gene therapy is another potential curative option with promising results, yet currently is offered only as part of a clinical trial.
Wiskott–Aldrich syndrome is a rare X-linked recessive inborn error of immunity first described in 1937. Classic WAS is characterized by the triad of thrombocytopenia with small platelets, recurrent infections due to combined immunodeficiency, and eczema.Citation1,Citation2 Furthermore, there is also an increased incidence of autoimmune conditions and lymphoid malignancies due to immune dysregulation. The WAS gene, which has 12 exons, is found on the short arm of the X chromosome at 11.22-23 locus and encodes a 502-amino-acid protein known as WAS protein (WASp). This cytosolic protein is expressed in myeloid, lymphoid, and hematopoietic stem cells (HSCs). WASp plays a key role in cytoskeletal remodeling and actin polymerization, as well as being a crucial regulator of the immunologic synapse between T and B cells.Citation3,Citation4 In addition, T regulatory function, NK cell cytotoxicity, monocyte chemotaxis, and dendritic cell shape have all been found to be aberrant in WAS patients.Citation5–Citation7 To date, more than 300 different WAS mutations have been discovered, with nine mutational hot spots accounting for around one-third of the overall number of mutations observed.Citation8
The global prevalence of WAS is estimated to be 1–10 males per million. As an X-linked disease, it usually affects males. Females are often obligate carriers; however, they can be affected by skewed lyonization, Turner syndrome, or a deleterious mutation on the paternal X chromosome.Citation3
Structure and Function of WAS Protein
WASp has three distinct functional domains: a pleckstrin homology domain that binds phosphatidylinositol bisphosphate; a Cdc42-binding domain; and a 70-amino-acid conserved verprolin-homology domain that forms the actin-binding area and is important for actin cytoskeleton control. WASp is a downstream effector of Cdc42, which is a small GTPase that regulates actin polymerization and cytoskeletal structure. The interaction of WASp with the Arp 2/3 complex is required for Cdc42-dependent actin assembly.Citation9–Citation11
Clinical Features
Although classic WAS is usually diagnosed within the first few months to a few years of life, diagnosis of milder cases can be delayed much farther into adulthood.Citation1,Citation12 The classical triad of microthrombocytopenia, recurrent infections and eczema is seen only in about a quarter of patients.
Thrombocytopenia
Thrombocytopenia is one of the first clinical signs of WAS. It is seen in nearly 80% of the patients at the time of diagnosis and about half of them have severe thrombocytopenia (platelet count <20,000/µL). The bleeding manifestations might range from non-life-threatening (petechiae, purpura, hematoma, epistaxis) to severe bleeding (intestinal and intracranial).Citation1,Citation13 While newborns present with bloody diarrhea/bleeding from the umbilical stump or bleeding after circumcision, the most common bleeding symptoms noticed in older children include cutaneous and gastrointestinal (GI) bleeding. Bleeding-related deaths occurred in 4–10% of the patients, with serious consequences observed with GI and intracranial bleeding.Citation1,Citation12 Thrombocytopenia occurs regardless of the severity of the mutation, which is attributed to the instability of the mutated WASp in platelets.Citation14 Normal mean platelet volume (MPV) is 7.2–11.7 fL and in WAS it is usually <5 fL. Micro platelets, considered the hallmark of WAS, were found only in 53% of the patients as reported by Sullivan et al.Citation1 There are anecdotal reports of WAS presenting with normal or even increased MPV, particularly in the context of autoimmunity, splenectomy, and post platelet transfusion.Citation15–Citation17 WAS can be misdiagnosed as immune thrombocytopenia (ITP), especially when thrombocytopenia is the only presenting symptom.Citation18 Thrombocytopenia in WAS is multifactorial and is attributed to defective platelet production from the megakaryocytes and splenic destruction involving both immune and non-immune mechanisms.
Immunodeficiency
The PID portion manifests with failure to thrive and recurrent infections due to combined immunodeficiency. The severity of immune deficiency may vary from family to family and sometimes can be life-threatening. Infections include bacterial (sinopulmonary infections, skin abscess, sepsis, meningitis), viral (herpes and severe hemorrhagic varicella), invasive fungal infections (candida), and opportunistic infections (Pneumocystis jirovecii).Citation12,Citation19 T cell defects are characterized by both qualitative and quantitative abnormalities. Though at birth T cell numbers are normal, progressive apoptosis leads to T lymphopenia by 6–8 years of age. Nearly half of the patients have an abnormal proliferative response to mitogens indicative of T cell qualitative abnormalities. B cell defects manifest with abnormal immunoglobulin profile, which includes low IgM, high IgA, and high IgE levels. IgG levels are usually normal, although the ability to mount sufficient antibody response to polysaccharide antigens and certain peptide antigens, including diphtheria, tetanus, and Hib vaccine, is decreased. This abnormal antibody response is a direct consequence of defective T cell and B cell interaction, resulting in failure of memory B cells to isotype switch. Abnormalities of NK cell function and phagocytic cells have also been reported. Defective NK cell cytotoxicity has been attributed to the increased risk of viral infections and malignancy. Functional defects in phagocytic cells due to defective chemotaxis, degranulation, and podosome assembly have been described, which are attributed to impaired intraphagosomal microbial killing.Citation9 Abnormal newborn screening assays for severe combined immunodeficiency can identify a small fraction of WAS, leading to early diagnosis before the onset of clinical symptoms.Citation20
Eczema
Eczema roughly affects three-quarters of WAS patients, with about half of those suffering from severe refractory eczema. XLT patients have milder symptoms due to residual WASp expression. Associated food allergies and secondary bacterial infections have been associated with severe exacerbations, which tend to improve with dietary modification and antibiotics. Although the exact cause of eczema in WAS patients is uncertain, a skewed Th2 cytokine profile resulting in elevated IgE levels and ectopic dermal location of Langerhan’s cell due to aberrant cell trafficking is proposed.Citation9,Citation21
Autoimmune Manifestations
Autoimmune manifestations were reported in 26–72% of WAS patients in various studies.Citation1,Citation4,Citation12,Citation22–Citation24 The most common autoimmune manifestation described in WAS patients is autoimmune hemolytic anemia (AIHA). In a cohort of 55 patients, Dupuis et al found that 36% had AIHA, 29% had arthritis, 25% had autoimmune neutropenia, 25% had vasculitis [skin (22%) and cerebral vasculitis (7%)], 9% had inflammatory bowel disease, and 3% had renal disease.Citation24 Other reported manifestations include autoimmune thrombocytopenia, IgA nephropathy, and a higher incidence of food and drug allergies.Citation23,Citation25 Autoimmunity has been described as an independent poor prognostic marker with a predisposition to malignancy.Citation1 The mechanism of autoimmunity is correlated with impaired Treg and B cell function. Treg cells play a vital role in the prevention of autoimmunity by modulating T-effector cells, which promote autoimmunity. Treg cells in WAS patients have an impaired ability to suppress activated T-effector cells, leading to loss of self-tolerance and autoimmunity.Citation2 Dupuis et al found a substantial correlation between high IgM levels and the development of autoimmunity. In their patient population, 90% of those with elevated IgM developed AIHA, in comparison to none with low IgM levels.Citation24 Also, abnormal NKT cell (cells possessing properties of NK and T cells) function has been described to play a role in autoimmunity due to the production of autoreactive B cells.Citation26
Malignancies
Hematolymphoid malignancies are the most common cancers associated with WAS. Adolescents and young adults are more likely to be affected than children. Sullivan et al found a 13% (21 of 154 patients) malignancy rate in their cohort. Hematologic malignancies were the most common, and the only three other non-hematologic malignancies reported were glioma, acoustic neuroma, and testicular carcinoma. The mean age of development of cancers in their patient cohort was 9.5 years. Due to its aggressive character, malignancy in the setting of WAS was associated with death in nearly all patients in this cohort.Citation1 Perry et al found that the relative risk of cancer was 100 times higher in the WAS group than in the general population, and the risk increased with age.Citation27 Lymphoid malignancies in WAS occur due to loss-of-function mutations, whereas acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) in X-linked neutropenia (XLN) occur due to gain-of-function mutations.Citation28 Non-Hodgkin lymphomas (NHL), especially B cell lymphomas, have a higher incidence than Hodgkin Lymphoma (HL) in WAS patients.Citation29 EBV-associated lymphoproliferative disorders (LPD) are frequently seen in patients receiving immunosuppressive therapy after organ transplantation but they can also occur in IEIs, including WAS.Citation12,Citation30
N-WASP, a protein closely related to WASp that is expressed primarily in neural cells, has 50% sequence homology to WASp and is also thought to play a role in the development of malignancies.Citation33 N-WASp is also an actin regulatory protein with a role in cytoskeletal reorganization, mainly in the actin-based filopodia in mammalian cells. N-WASp has a crucial role in cancer invasion as elucidated in various studies. This occurs through Arp 2/3-complex-mediated actin polymerization and matrix metalloproteinase (MMR) dependent mechanisms. Liu et al described the potential role of N-WASp as a prognostic factor in clear cell renal carcinoma. They found low expression of N-WASp correlated with poor histologic grades; however, high expression correlated with poor survival.Citation31 Various other independent observers have described the role of N-WASp and its prognostic significance in endometrial cancer, hepatocellular carcinoma, breast cancer metastasis, and esophageal squamous cell carcinoma.Citation32–Citation34
Atypical Presentations
A subset of patients with WAS rather present with atypical manifestations, and unless, already been diagnosed with WAS or there is a strong family history of WAS, this can pose diagnostic challenge. There are few published case reports of occurence of aortic aneurysms in WAS diagnosed patients during follow-up, needing immediate surgical repair.Citation35,Citation36 Pellier et al reported the occurrence of aortic aneurysms in 5 out of 38 patients (13%) between the ages of 10–16 years.Citation37 Barutcu et al reported a 7-year-old boy managed as chronic ITP who was diagnosed with WAS after noticing an aortic aneurysm.Citation38 The pathophysiology of WAS-associated vasculitis remains unclear but is attributed to immunoglobulin A and E deposition in the vessel wall leading to chronic inflammation. Aneurysmal rupture is associated with a high risk of mortality, and hence some experts recommend periodic echocardiography and magnetic resonance imaging for surveillance due to higher likelihood of its occurrence in WAS.
He et al reported a toddler boy with WAS presenting with extensive subcutaneous bleeding, thrombocytopenia and isolated prolonged activated partial thromboplastin time (APTT). On further investigation, this patient had Factor VIII deficiency and an extremely high level of Factor VIII inhibitor, which led to the diagnosis of acquired hemophilia A.Citation39 Patil et al described a 3-month-old male infant who presented with poor feeding/oral ulceration, coomb’s positive hemolytic anemia and thrombocytopenia who was being managed as Evan’s syndrome, until the development of generalized eczematous rash and the genetic testing then revealed WAS mutation.Citation40 Severe generalized eczema when combined with bleeding manifestations and severe recurrent infections not limited to the skin should raise the suspicion for WAS as reported by Kumar et al.Citation41
Genotype–Phenotype Correlation
Using linkage and mutation analysis it was established that XLT is caused by mutations in the same gene as WAS. WASp gene mutations result in three distinct phenotypes: Classic WAS, milder XLT variant (characterized only by thrombocytopenia/mild eczema), and X-linked neutropenia (XLN) without any findings of WAS/XLT. Jin et al found a high genotype–phenotype correlation in their research of two large cohorts in North America and Europe.Citation5 Absent WASp expression (WASp-) leads to a more severe phenotype (WAS), whereas residual protein expression (WASp+) results in milder phenotypes (XLT/XLN).Citation8 Single nucleotide substitutions, small insertions, deletions, splice site mutations throughout the coding regions, and intron/exon junctions were the most often noted mutations in various studies.Citation9,Citation42 The most common mutations observed in WASp+ patients were missense mutations located within the PH domain (exons 1–3), whereas WASp- patients had nonsense mutations/deletions/insertions.Citation12,Citation43,Citation44 WASp activating mutations are thought to be the cause of XLN.Citation28 Among five common mutational “hot spots”, three of them (168 >T mutation, 290C>N/291G>N mutation, IVS6 +5g>a mutation) occurred predominantly in WASp+ (mild phenotype), whereas 665C> T mutation and IVS8+1 g mutations occurred in WASp- patients (severe phenotype) among unrelated families (P< 0.001).Citation5 Environmental factors and genetic determinants unrelated to WASp, on the other hand, are likely to influence the clinical phenotype, which could explain the variable WAS phenotypes seen in some families.Citation1,Citation45
Scoring Systems
The clinical scoring system uses five parameters, namely thrombocytopenia, the severity of eczema, infections, development of autoimmunity, and malignancy. Each component is given a score of 1 with a total score of 5. Patients with a classic triad obtain a score of 3–4, while those with autoimmunity or malignancy receive a score of 5.
Any score ≥3 indicates a severe phenotype, and those with a score of <3 indicates a milder phenotype/XLT, which is characterized by only thrombocytopenia with mild/no eczema, and absence/mild recurrent infections and scores anywhere between 0.5 and 2.5.Citation4,Citation19 A score of 0.5 is given for intermittent thrombocytopenia in the absence of other symptoms.Citation46 This classification helps predict which patients are likely to develop severe complications and would benefit from an early HSCT. Since the disease might not have fully evolved, this scoring system is less reliable in infants and young children (<2 years). Hence, this predictive model is not absolute and based on the clinical course of the individual patients, further evaluation for WASp expression and/or genetic testing is needed.
Diagnosis
The management of WAS begins with a proper diagnosis of the condition as clinical manifestations are extremely heterogeneous. The classic triad is seen only in about 15–27%.Citation1,Citation47 Because of the wide range of clinical presentations, the median age of diagnosis ranged from 1.75 to 24 months in various studies.Citation19,Citation25,Citation48 Recurrent infections or autoimmunity could be the presenting complaints in a small portion of patients. WAS should always be suspected in a male patient with thrombocytopenia and low mean platelet volume. Diagnostic tests include screening for T and B cell function, mutational analysis of DNA, and flow cytometry for WASp expression.
Treatment
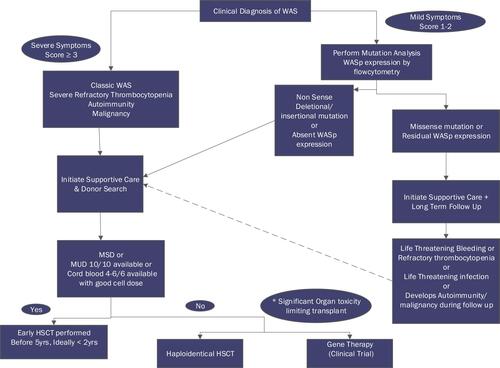
Depending on the severity, a variety of therapeutic options are available for patients with WAS. Potential treatment options include supportive care, splenectomy, hematopoietic stem cell transplantation (HSCT), and autologous HSC gene therapy. More than one of these modalities may be required for any one patient. WAS, being a complex and chronic disorder, is best managed by a multidisciplinary team including stem cell transplant physicians, pediatric immunologists, dermatologists, dietitians, geneticists, and social workers. The standard of care for classic WAS is early transplant before the onset of severe infections, autoimmunity, and malignancy. Treatment decision based on the clinical symptoms, genetic mutation, and WASp expression is described in .
Figure 1 Treatment decision algorithm for WAS/ XLT based on the severity of clinical symptoms, type of genetic mutation, and WASp expression. For patients with classic WAS with a suitable MSD/MUD donor or CB 5-6/6 or 4/6 with a good cell dose, an early transplant should be performed. In the absence of a suitable matched donor, a Haploidentical transplant or gene therapy trial should be discussed. XLT patients with severe thrombocytopenia or mutation suggestive of a severe phenotype and absent WASp expression should also be transplanted.
Supportive Care
Supportive care is critical in the management of patients with WAS/XLT. This includes management of bleeding symptoms, initiating antimicrobial prophylaxis and immunoglobulin (IgG) replacement for classic WAS and XLT patients with recurrent infections, and appropriate management of autoimmune manifestations and malignancy. Patients with severe eczema should be referred to dermatology and dietitian for appropriate management of food allergy. This is crucial due to increased infection risk from the central line and immunosuppression caused by the transplant. XLT requires little supportive care when compared to classical WAS. Supportive care for patients is summarized in .
Table 1 Supportive Care in WAS
Alternative Therapies for Thrombocytopenia
Role of Splenectomy
Splenectomy has been shown to normalize platelet counts and lower the risk of major bleeding in WAS/XLT. In one study due to normalization of platelet counts, splenectomized patients had a much longer median survival than non-splenectomized patients, with a median survival of 25 years compared to 5 years in patients who did not undergo transplant.Citation49 However, splenectomy raises the risk of overwhelming sepsis, especially in the post-transplant period despite being on prophylactic antibiotics. Various studies have also found no reduction in the risk of autoimmune and lymphoproliferative disorders after HSCT in splenectomized patients.Citation50,Citation51 Lifelong antibiotic prophylaxis is recommended for WAS/XLT patients who undergo splenectomy. As a result, the current trend is to avoid splenectomy, especially in patients who will undergo transplants.
Low-Dose Interleukin-2 (IL-2)
Low-dose IL-2 was employed as an immunostimulant treatment in WAS/XLT patients in Phase 1 clinical research at a level of 0.5 million U/m2/day. There was a statistically significant rise in platelet count, as well as high T, B, and NK cell numbers and T regulatory cell percentages. More research is needed, however, to prove efficacy.Citation52
Thrombopoietin Mimetics
Eltrombopag has been explored in patients with hereditary thrombocytopenia, including WAS/XLT, and has shown a slight rise in platelet count. Zaninetti et al in their Phase II clinical trial enrolling patients with various inherited thrombocytopenia including WAS/XLT, treated patients with a 3- to 6-week course of eltrombopag. The pretreatment platelet count of the cohort was 40,000/µL and post-treatment, 11 out of 23 patients (47.8%) showed a major response (platelet count >100,000/µL) and 10 (43.5%) showed a minor response (platelet count at least twice the baseline value) and 2 (8.7%) had no response. Eltrombopag was well tolerated without any major side effects.Citation53
Eltrombopag was used as a bridge to transplant in a patient with early-onset WAS with severe refractory thrombocytopenia with a high frequency of clinical bleeding. The initial starting dose of eltrombopag used was 0.8 mg/kg/day and increased up to a maximum of 5 mg/kg/day. The patient benefitted with a significant reduction in clinical bleeding symptoms with complete elimination of platelet transfusion reducing the risk of alloimmunization.Citation54
Gerrits et al investigated the effect of eltrombopag on platelet count and platelet activation in WAS/XLT patients in a clinical trial enrolling nine WAS/XLT patients and eight age-matched controls. In this study, eltrombopag had favorable effects on increasing platelet count but not platelet activation in WAS/XLT, the reason being micro platelets. Platelet function as measured by agonist-induced surface GPIIb-IIIa and P-selectin activation was proportional to the platelet size in WAS/XLT patients.Citation55
Autoimmunity and Malignancy
AIHA is treated with corticosteroids. Other immunosuppressive medications, such as IVIG, rituximab, cyclophosphamide, azathioprine, and calcineurin inhibitors, have been utilized to treat autoimmune diseases linked to WAS. The use of immunosuppressive drugs to treat autoimmunity could potentially increase the risk of infection in WAS patients. In several trials, definitive therapy with HSCT resulted in the resolution of autoimmunity.Citation4 Treatment for various cancers is similar to that of other pediatric oncology patients.
Definitive Therapy
Hematopoietic Stem Cell Transplantation in WAS
The first successful transplant for WAS was performed in 1968 on a 2-year-old boy.Citation56 Since then, allogeneic HSCT has emerged as the most reliable curative treatment for WAS as it can correct both hematologic and immunologic abnormalities. Early referral to transplant and suitable donor search should be initiated immediately following diagnosis. HLA typing of the patient and all potential family members should be implemented. In the absence of a suitable family donor, matched unrelated donor search through a worldwide registry should be initiated. Various published studies on transplants along with salient features are illustrated in .
Table 2 Allogenic HSCT in WAS and Their Salient Features
Indications for Transplant
The decision to transplant is usually made based on the clinical severity, the type of genetic mutation, and residual WASp expression. HSCT is recommended for all patients with classic/severe WAS (Clinical score ≥3) and those with severe refractory thrombocytopenia due to the risk of life-threatening bleeding. Autoimmunity or malignancy (clinical score 5) are two other high-risk conditions that require transplantation. In young children in whom disease manifestation is yet incomplete, the presence of severe genetic mutation and/or absence of WASp expression should guide towards transplant, as the natural course of the disease is expected to worsen over time. Imai et al in their study demonstrated a correlation of WASp expression to survival, where WASp – (n=23) patients had a lower 10-year overall survival (OS) than WASp+ (n=27) individuals (76% vs 92.3%).Citation12
Unlike classic WAS, clear recommendations on the role and timing of HSCT in XLT are lacking. Albert et al found that, despite good OS rates, event-free survival (EFS) was unsatisfactory in a long-term outcome study of XLT patients. XLT individuals suffered major bleeding (13.9%), life-threatening infections (6.9%), autoimmunity (12.1%), and cancer (5.2%) during follow-up, according to the same study.Citation63 As a result, if MSD is available, HSCT should be considered for XLT patients.
Timing of Transplant
Multiple studies have consistently found that transplants performed at a younger age (under 5 years) had superior OS rates.Citation51,Citation64 Burroughs et al found that in their cohort of 129 patients, the 5-year OS rate was 94% vs 66% with P=0.0008 for patients transplanted <5 years vs >5 years, respectively.Citation4 Also, they reported intensity of conditioning regimen, donor type, hematopoietic stem cell source, HSCT year, WAS score, and WASp expression were not significantly associated with OS.Citation4 On the contrary, Moratto et al described transplants performed after the year 2000 as having improved survival for all donor types.Citation51 Transplant at an older age may be complicated by the risk of primary disease progression with development of autoimmunity or cancer, viral reactivation, and a higher prevalence of graft versus host disease (GVHD).Citation64–Citation66 Whereas, transplant in infants should be proceeded with caution due to the potential disadvantage of toxicity of the conditioning regimen and variation in drug metabolism at such a young age.
Donor Type and Graft Source
A matched sibling donor (MSD) is the preferred donor for HSCT with excellent 5-year Overall Survival (OS) 90–95% and event-free survival (EFS) of 88%.Citation22,Citation50,Citation64 Historically, inferior outcomes were reported in the setting of matched unrelated donors (MUD) and mismatched related donors (MMRD) when compared to MSD. Filipovich et al in a cohort of 170 patients, studied between 1968 and 1996 in a collaborative study of International Bone Marrow Transplant Registry (IBMTR) and National Marrow Donor Program (NMDP) reported a 5-year OS of 87% with MSD, 52% with other related donors and 71% with unrelated donors. This excellent outcome with unrelated donors was reported when age at transplant was <5 years; however, patients >5 years had a poor outcome.Citation57 Contrary to this study, recent literature shows improved transplant outcomes over time for all donor types.Citation4,Citation50 This is attributed to better donor selection and a wider worldwide donor base, use of less toxic conditioning regimen, effective GVHD management, and advances in prevention and treatment of infection.Citation51
Alternative donor transplants including umbilical cord blood (UCB) and mismatched related/haploidentical transplants were demonstrated to have poor outcomes according to earlier studies.Citation50,Citation51 However, in the last decade, better cord selections and effective measures to overcome graft failure/rejection and GVHD have paved the way for successful use of alternative donors in WAS boys who lack suitable matched donors.Citation4,Citation50,Citation51 In a Primary Immunodeficiency Disease Treatment Consortium (PIDTC) report conducted between 2005 and 2015, Burroughs et al reported use of CB grafts in 39 of 129 patients (30%) and reported an excellent 5-year OS of 90%. Shekhovtsva et al published results for UCB outcomes on 90 patients using myeloablative (MAC) conditioning and anti-thymocyte globulin and reported five-year OS rates of 75% and an EFS of 70%. Improved outcomes with CB in this study have also been attributed to better cord selection, higher CD34 cell dose, and improved supportive care.
Before the year 2000, T cell-depleted haploidentical or mismatched unrelated grafts were reported to be associated with high transplant-related mortality (TRM) of 38–55%, which was attributed to graft failure, GVHD, and infectious complications.Citation46 Improved outcomes reported in the recent era are due to the use of or post-transplant cyclophosphamide (PT-Cy) or T cell depletion bearing αβ receptor implicated in causing GVHD, and CD19-positive cells harboring EBV.Citation62,Citation67 Balashov et al reported a dramatic decrease in the incidence of GVHD and TRM in a haploidentical transplant with T cell receptor (TCR)αβ+/CD19+ graft depletion with a reported OS of 88.9%. However, mixed myeloid donor chimerism and secondary graft rejection (graft rejection and severe thrombocytopenia) were reported in 7 out of 18 patients in the current series. Hence, in patients undergoing MUD/Haplo transplant with TCR αβ+/CD19+ graft depletion, the same investigators proposed the addition of G-CSF (10 μg/kg/day for 5 days starting on day −8) and plerixafor (240 μg/kg/day for 3 days starting on day −6) to the conditioning regimen, in a clinical trial and demonstrated full donor chimerism with OS rates of 93.8% without any graft dysfunction.Citation62 Another recent study by Elfeky et al reported 100% OS in 34 patients including alternative donor transplants with a median follow-up of 63 months.Citation61 Though very promising, limitations of TCR αβ+ depletion include high cost and restricted availability, making PT-Cy a realistic choice for GVHD prophylaxis in alternative donor transplant.Citation68
Bone marrow (BM) as a graft source is preferred due to the high risk of chronic GVHD with peripheral blood stem cells. A fully matched graft (either BM or PBSC) is a 10/10 match at HLA-A, -B, -C, -DRB1, and -DQB1 at the antigen level for MSD and allele level for MUD. For cord blood, 6/6 is considered a full match at HLA-A and -B at the antigen level and -DRB1 at the allele level.Citation4
Conditioning Regimen
The first patient who underwent HSCT for WAS received minimal conditioning and demonstrated donor-derived T-cell reconstitution but failed to achieve donor-derived myeloid reconstitution. However, a second transplant from the same donor following a myeloablative conditioning regimen resulted in successful platelet engraftment. These initial transplant experiences highlighted that WAS patients have a selective advantage for WASp expression in lymphoid lineage compared to platelets, resulting in early T cell reconstitution and no platelet reconstitution unless both myeloablative and immune-ablative conditioning regimen is used. Conventional myeloablative conditioning regimens used include Busulfan and Cyclophosphamide. To reduce the toxicity associated with the conditioning regimen, reduced-dose busulfan and treosulfan-based regimens are increasingly being used. One of the primary problems with WAS post-transplant is mixed donor chimerism, which was highest with the reduced-intensity conditioning (RIC) regimens.Citation4,Citation50,Citation69 Furthermore, even with myeloablative conditioning (MAC) regimens, a substantial risk of mixed donor chimerism exists (19–30%), especially with MMRD. The degree of mixed donor chimerism in the post-transplant setting is a significant factor affecting platelet reconstitution. Hence, many experts in the field still recommend the use of MAC regimens due to higher frequencies of achieving full donor chimerism and better T-cell engraftment.Citation4,Citation51,Citation59
Since WAS patients exhibit robust T Cell function compared to SCID patients, most protocols now include serotherapy (anti-thymocyte globulin [(ATG)/alemtuzumab]) in their conditioning regimens. Elfeky et al used RIC based on reduced-dose busulfan or treosulfan along with serotherapy with alemtuzumab/rabbit ATG and showed 100% survival with a median follow-up of 63 months in their cohort of patients with most patients receiving alternative donors.Citation61 Treosulfan/lower-dose busulfan (area under the curve 65 mg/L/h), fludarabine, and serotherapy are currently the most widely employed reduced toxicity regimen.Citation70
Nonmyeloablative conditioning was used by Burroughs et al for patients in whom infection/organ dysfunction precluded the use of MAC. Although stable donor engraftment and low early mortality were observed among those patients, a higher rate of GVHD was noted.Citation71
Post-Transplant Complications
Most transplant-related complications occur within the first year after transplant and include mixed chimerism, infectious risk, autoimmunity, and malignancy, GVHD, TRM, and late effects such as infertility, second malignancies, endocrine issues, and organ dysfunction.
Mixed Chimerism
Mixed donor chimerism is a particular problem in patients with WAS in the post-transplant period. The reported incidence of mixed donor chimerism in various studies has been ~19–50%.Citation50,Citation51,Citation69 Mixed donor chimerism results in poor platelet reconstitution, subnormal lymphocyte counts, and autoimmune illness.Citation22,Citation50,Citation51 According to Moratto et al, donor chimerism in the T-cell, B-cell, and Myeloid compartment was classified into four groups: full (>95% donor cells), high (>50–95%), low (5–50%), and null (<5%) chimerism. In their cohort, 72.1% of the patients achieved full donor chimerism in all three compartments, 27.9% mixed chimerism in at least one of the compartments, low or null donor chimerism was more common in myeloid cells (16.5%) in comparison to B cells (7.4%) and T cells (3.2%). Although low donor chimera was seen in the first year after HSCT, improved donor chimerism with stabilization was reported in a significant proportion of patients over time.Citation51 Platelet reconstitution after HSCT is linked to the degree of myeloid donor chimerism, with higher platelet counts achieved in full donor chimerism compared to low-level chimerism.Citation4 Type of conditioning was the major determinant of mixed donor chimerism, with RIC linked to a higher likelihood than MAC.
Donor myeloid chimerism <50% was associated with higher rates of platelet transfusion dependence and subsequent graft failure.Citation51,Citation72 One other mechanism for a low degree of donor myeloid chimerism is the emergence of autoimmune thrombocytopenia in the post-transplant period.Citation72 In cases of incomplete reconstitution, close monitoring of the donor chimerism is critical, and therapies such as stem cell boost, donor lymphocyte infusion, or a second transplant may be required.
Post-Transplant Autoimmunity
The incidence of post-transplant autoimmunity usually occurs within 2 years after HSCT and has been reported to be 20%.Citation50 Pre-transplant autoimmunity was not a risk factor for post-transplant autoimmunity. Although earlier studies reported mixed donor chimerism after MUD or MMRD HSCT as an independent risk factor to develop de novo post-transplant autoimmunity, subsequent studies failed to reproduce the same results.Citation4,Citation22,Citation50,Citation51,Citation61 In a recent study, Burroughs et al reported an increased incidence of autoimmunity with mixed myeloid chimerism at 6 months post-transplant; however, this was not observed on long-term follow-up. Donor type is a significant consideration as post-transplant autoimmunity was seen in none of the MSD, unlike 23% of unrelated and 9% CB grafts.Citation4
Autoimmune cytopenias (hemolytic anemia, thrombocytopenia, and neutropenia) are the predominant manifestation of autoimmunity in the post-transplant period, with a reported incidence varying between 14% and 72%.Citation12,Citation50,Citation69,Citation73
GVHD
Sophisticated HLA matching technology, allograft manipulation in alternative donor transplants, and with appropriate GVHD prophylaxis, the incidence of acute GVHD has decreased in WAS transplanted patients. The cumulative incidence of day 100 chronic GVHD as reported by Burroughs et al was 17%, and there was no difference in the incidence of acute/chronic GVHD based on the different graft types.Citation4
Infections
WAS transplanted patients are at risk for serious, as well as fatal, infections, even 2 years after transplant. Ozashin et al demonstrated that risk of severe infections correlated with splenectomy, and strongly advocated against splenectomy in patients who are transplant candidates/transplant recipients.Citation50 Shin et al reported viral reactivation in 84% (26/31 patients) of transplant recipients using viral polymerase chain reaction (PCR) tests. Age at transplant was a significant predictor in their study, which demonstrated patients transplanted <2 years had 71% viral reactivation vs 100% in those transplanted >2 years.Citation22 Periodic surveillance with quantitative PCR, preventive measures and aggressive treatment of infections are crucial in the post-transplant period.
Post-Transplant Malignancy
The true incidence of post-transplant malignancy is unknown due to limited published results. Kamani et al in their CIBMTR study on malignancies after HSCT in primary immunodeficiency disorders (PIDs) reported the highest incidence of malignancy of 3.3% (n=360 patients) for WAS when compared to 2.3% for other PIDs. Various reported malignancies included lymphoproliferative disorders (LPD) [mainly, EBV-related post-transplant lymphoproliferative disorder (PTLD)], Immunoblastic B-cell lymphoma and large-cell lymphoma, diffuse large B-cell lymphoma (DLBCL) high-grade NHL, myelodysplasia and AML.Citation3,Citation22,Citation50,Citation51,Citation74 Significant risk for development of LPD was the use of T cell-depleted graft as demonstrated by the fact that the median time to LPD from HSCT was 3 months.Citation74,Citation75
Late Effects
While the overall incidence of late effects after HSCT for WAS is lower than for SCID, Ozashin et al found a higher incidence (20%) of late complications and irreparable damage with sequela of those who survived beyond 2 years with a 7-year EFS of 75%. Long-term side effects from conditioning regimens, chronic GVHD, veno-occlusive disease, endocrine complications, and hampered growth and development are of main concern. Unique challenges of transplanting an immunodeficiency disorder include systemic distribution of some of these genetic defects and incomplete understanding of the natural history of these defects.
Gene Therapy for WAS
Gene therapy (GT) using viral vectors to treat WAS has the potential to become a promising alternative for patients without a suitably matched donor and/or patients with disease-related comorbidities that increase the risk for transplant-related mortality. The ex-vivo transduction of autologous hematopoietic stem cells with lentiviruses or retroviruses can be used to restore normal WASp to cells.Citation76
The first gene therapy clinical trial for WAS started in 2006 in Hanover, Germany. The trial used a Moloney Leukemia virus-derived γ-retroviral vector. Ten patients were treated with peripheral blood mononuclear cells after receiving GCSF ± plerixafor. The conditioning regimen consisted of Busulfan 8mg/kg followed by the infusion of 2.9×106 to 24.9×106 CD34+ cells/kg. Although nine out of ten patients achieved immune reconstitution and correction of WASP expression with the improvement of clinical status and WAS scores, this trial had an unacceptably high risk of insertional mutagenesis, with activation of several proto-oncogenes leading to T-ALL in six patients (488–1813 days after GT) and one patient with AML 1165 days after GT. Blood analysis of these patients showed clusters of integration sites near LMO2, CCND2, MDS1/EVI1 genes.Citation77,Citation78
Subsequent clinical trials have used a self-inactivating lentiviral vector (SIN-LV) for WAS gene correction. Conditioning regimens for these trials included Busulfan (12mg/kg), and Fludarabine 120mg/m2. Target busulfan AUC differed between trials from 60 to 82. The trial conducted in Milan used a lower dose of busulfan (6.4–9.6 mg/kg with a target AUC of ~48), Fludarabine 60mg/m2, and anti-CD20 monoclonal antibody (mAb). To date, a total of 34 patients with WAS have been treated with LV-transduced autologous bone marrow or mobilized peripheral-derived CD34+ cells. Ninety-one percent of the patients survived and sustained multi-lineage engraftment of cells expressing the WASp with associated significant improvement of symptoms.Citation79–Citation81 Even in the European trial that included mostly patients with severe WAS (score 5 in 6 out 7 patients) gene therapy was well tolerated with evident clinical improvement. One patient died from multiagent-resistant septic shock 7 months after gene therapy. The surviving patients experienced resolution of severe eczema, recurrent infections, autoimmunity, and after 7 months, these patients did not require regular transfusions or the use of TPO mimetics. Immune reconstitution with normal absolute T cell counts was seen in four out of six patients. Despite showing an improvement in platelet size, platelet numbers remained below normal values for all patients, but no severe bleeding symptoms were reported.Citation79 In contrast, two out of the five patients treated in Boston did not show resolution of autoimmunity after gene therapy. Gene therapy for WAS using SIN-LV has not been associated with severe adverse events, including no evidence of the transformation of hematopoietic cells as was observed with the previous retroviral vectors despite vector integration adjacent to proto-oncogenes.Citation79,Citation80,Citation82
A recently published long-term analysis of seventeen patients treated with WASp SIN-LV OTL-103 at an Italian hospital reported that all surviving patients who achieved multilineage engraftment and sustained WASp expression in lymphocytes and platelets, sustained improvement in infection risk and bleeding events after 8 years post-treatment.Citation80 Salient features of HSCT versus gene therapy are highlighted in .
Table 3 HSCT Vs Gene Therapy
In summary, outcomes for WAS have significantly improved over the years with HSCT. Even with 90–100% survival reported in multiple studies, acute and long-term toxicity associated with transplant remains a concern.Citation3,Citation61 Gene therapy has demonstrated a better safety profile and can improve all features of WAS, despite limited long-term follow-up data. However, with the widespread availability and long-term safety data, gene therapy has the potential to become a standard therapeutic option.
Acknowledgments
Mandy Neudecker, Pediatric Librarian at Rainbow Babies and Children’s Hospital.
Disclosure
The authors report no conflicts of interest in this work.
References
- Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA. A multiinstitutional survey of the Wiskott-Aldrich syndrome. J Pediatr. 1994;125(6):876–885. doi:10.1016/S0022-3476(05)82002-5
- Massaad MJ, Ramesh N, Geha RS. Wiskott-Aldrich syndrome: a comprehensive review. Ann N Y Acad Sci. 2013;1285(1):26–43. doi:10.1111/nyas.12049
- Buchbinder D, Nugent DJ, Fillipovich AH. Wiskott-Aldrich syndrome: diagnosis, current management, and emerging treatments. Appl Clin Genet. 2014;7:55–66. doi:10.2147/TACG.S58444
- Burroughs LM, Petrovic A, Brazauskas R, et al. Excellent outcomes following hematopoietic cell transplantation for Wiskott-Aldrich syndrome: a PIDTC report. Blood. 2020;135(23):2094–2105. doi:10.1182/blood.2019002939
- Jin Y, Mazza C, Christie JR, et al. Mutations of the Wiskott-Aldrich Syndrome Protein (WASP): hotspots, effect on transcription, and translation and phenotype/genotype correlation. Blood. 2004;104(13):4010–4019. doi:10.1182/BLOOD-2003-05-1592
- Dupré L, Aiuti A, Trifari S, et al. Wiskott-Aldrich syndrome protein regulates lipid raft dynamics during immunological synapse formation. Immunity. 2002;17(2):157–166. doi:10.1016/S1074-7613(02)00360-6
- Blundell MP, Worth A, Bouma G, Thrasher AJ. The Wiskott-Aldrich syndrome: the actin cytoskeleton and immune cell function. Dis Markers. 2010;29(3–4):157–175. doi:10.3233/DMA-2010-0735
- Moratto D, Giliani S, Notarangelo LD, Mazza C, Mazzolari E, Notarangelo LD. The Wiskott-Aldrich syndrome: from genotype-phenotype correlation to treatment. Expert Rev Clin Immunol. 2007;3(5):813–824. doi:10.1586/1744666X.3.5.813
- Ochs HD, Thrasher AJ. The Wiskott-Aldrich syndrome. J Allergy Clin Immunol. 2006;117(4):725–738. doi:10.1016/j.jaci.2006.02.005
- Symons M, Derry JMJ, Karlak B, et al. Wiskott-Aldrich syndrome protein, a novel effector for the GTPase CDC42Hs, is implicated in actin polymerization. Cell. 1996;84(5):723–734. doi:10.1016/S0092-8674(00)81050-8
- Stewart DM, Tian L, Nelson DL. Mutations that cause the Wiskott-Aldrich syndrome impair the interaction of Wiskott-Aldrich syndrome protein (WASP) with WASP interacting protein. J Immunol. 1999;162(8):5019–5024.
- Imai K, Morio T, Zhu Y, et al. Clinical course of patients with WASP gene mutations. Blood. 2004;103(2):456–464. doi:10.1182/blood-2003-05-1480
- Ochs HD. The Wiskott-Aldrich syndrome. IMA. 2002;4:379–384.
- Bosticardo M, Marangoni F, Aiuti A, Villa A, Roncarolo MG. Recent advances in understanding the pathophysiology of Wiskott-Aldrich syndrome. Blood. 2009;113(25):6288–6295. doi:10.1182/blood-2008-12-115253
- Patel PD, Samanich J, William Mitchell DM, Manwani D. A unique presentation of Wiskott–Aldrich syndrome in relation to platelet size. Pediatr Blood Cancer. 2011;56(7):1127–1129. doi:10.1002/pbc.22920
- Skoric D, Dimitrijevic A, Cuturilo G, Ivanovski P. Wiskott-Aldrich syndrome with macrothrombocytopenia. Indian Pediatr. 2014;51(12):1015–1016. doi:10.1007/s13312-014-0550-5
- Mazumdar J, Kanjilal S, Das A. Wiskott-Aldrich syndrome with normal-sized platelets in an eighteen-month-old boy: a rare mutation. J Pediatr Rev. 2015;3(2):10–13. doi:10.17795/jpr-417
- Bader-Meunier B, Proulle V, Trichet C, et al. Misdiagnosis of chronic thrombocytopenia in childhood. J Pediatr Hematol Oncol. 2003;25(7):548–552. doi:10.1097/00043426-200307000-00010
- Suri D, Rikhi R, Jindal AK, et al. Wiskott Aldrich Syndrome: A Multi-Institutional Experience From India. Front Immunol. 2021;12:627651. doi:10.3389/fimmu.2021.627651
- Borte S, Fasth A, von Döbeln U, Winiarski J, Hammarström L. Newborn screening for severe T and B cell lymphopenia identifies a fraction of patients with Wiskott–Aldrich syndrome. Clin Immunol. 2014;155(1):74–78. doi:10.1016/J.CLIM.2014.09.003
- Trifari S, Sitia G, Aiuti A, et al. Defective Th1 cytokine gene transcription in CD4 + and CD8 + T cells from Wiskott-Aldrich syndrome patients. J Immunol. 2006;177(10):7451–7461. doi:10.4049/jimmunol.177.10.7451
- Shin CR, Kim MO, Li D, et al. Outcomes following hematopoietic cell transplantation for WiskottAldrich syndrome. Bone Marrow Transplant. 2012;47(11):1428–1435. doi:10.1038/bmt.2012.31
- Chen N, Zhang ZY, Liu DW, Liu W, Tang XM, Zhao XD. The clinical features of autoimmunity in 53 patients with Wiskott–Aldrich syndrome in China: a single-center study. Eur J Pediatr. 2015;174(10):1311–1318. doi:10.1007/s00431-015-2527-3
- Dupuis-Girod S, Medioni J, Haddad E. Autoimmunity in Wiskott-Aldrich syndrome: risk factors, clinical features and outcome in a single center cohort of 55 patients. Pediatrics. 2003;111(5):e622–e627. doi:10.1542/peds.111.5.e622
- Lee PPW, Chen TX, Jiang LP, et al. Clinical and molecular characteristics of 35 Chinese children with Wiskott-Aldrich syndrome. J Clin Immunol. 2009;29(4):490–500. doi:10.1007/s10875-009-9285-9
- Mars LT, Araujo L, Kerschen P, et al. Invariant NKT cells inhibit development of the Th17 lineage. Proc Natl Acad Sci. 2009;106(15):6238–6243. doi:10.1073/pnas.0809317106
- Perry GS, Spector BD, Schuman LM, et al. The Wiskott-Aldrich syndrome in the United States and Canada (1892–1979). J Pediatr. 1980;97(1):72–78. doi:10.1016/S0022-3476(80)80133-8
- Keszei M, Snapper SB, Westerberg LS, et al. Constitutive activation of WASp in X-linked neutropenia renders neutrophils hyperactive graphical abstract find the latest version: constitutive activation of WASp in X-linked neutropenia renders neutrophils hyperactive. J Clin Invest. 2018;128(9):4115–4131. doi:10.1172/JCI64772
- Cotelingam JD, Witebsky FG, Hsu SM, Blaese RM, Jaffe ES. Malignant lymphoma in patients with the Wiskott-Aldrich syndrome. Cancer Invest. 1985;3(6):515–522. doi:10.3109/07357908509039813
- Yoshida K, Minegishi Y, Okawa H, et al. Epstein-Barr virus-associated malignant lymphoma with macroamylasemia and monoclonal gammopathy in a patient with Wiskott-Aldrich syndrome. Pediatr Hematol Oncol. 1997;14(October 1995):1–5.
- Liu GH, Chen J, Ji ZG, Zhou L. Expression of neural Wiskott-Aldrich syndrome protein in clear cell renal cell carcinoma and its correlation with clinicopathological features. Urol Int. 2015;95(1):79–85. doi:10.1159/000365595
- Huang L, Lian J, Chen X, Qin G, Zheng Y, Zhang Y. WASH overexpression enhances cancer stem cell properties and correlates with poor prognosis of esophageal carcinoma. Cancer Sci. 2017;108(12):2358–2365. doi:10.1111/cas.13400
- Martin TA, Pereira G, Watkins G, Mansel RE, Jiang WG. N-WASP is a putative tumour suppressor in breast cancer cells, in vitro and in vivo, and is associated with clinical outcome in patients with breast cancer. Clin Exp Metastasis. 2008;25(2):97–108. doi:10.1007/s10585-007-9120-8
- Jin KM, Lu M, Liu FF, Gu J, Du XJ, Xing BC. N-WASP is highly expressed in hepatocellular carcinoma and associated with poor prognosis. Surg (United States). 2013;153(4):518–525. doi:10.1016/j.surg.2012.08.067
- Bernabeu E, Josa M, Nomdedeu B, et al. One-step surgical approach of a thoracic aortic aneurysm in Wiskott-Aldrich syndrome. Ann Thorac Surg. 2007;83(4):1537–1538. doi:10.1016/j.athoracsur.2006.10.005
- Wood G, Booth K, Khan Z, Biss T, Roysam C, Dark J. Descending aortic aneurysm in Wiskott-Aldrich syndrome: options for repair. Asian Cardiovasc Thorac Ann. 2017;25(9):635–637. doi:10.1177/0218492317738386
- Pellier I, Girod SD, Loisel D, et al. Occurrence of aortic aneurysms in 5 cases of Wiskott-Aldrich syndrome. Pediatrics. 2011;127(2):e498–e504. doi:10.1542/peds.2009-2987
- Barutcu A, Goksel Leblebisatan SL, Metin Cil HI, Sasmaz FD, Sasmaz HI, Demir F. A case with Wiskott-Aldrich syndrome and ascending aorta aneurysm. J Pediatr Hematol Oncol. 2021;43:774–776. doi:10.1097/MPH.0000000000001932
- He T, Cao K, Qi Z, et al. Acquired hemophilia A in Wiskott–Aldrich syndrome. J Clin Immunol. 2021;41:1119–1122. doi:10.1007/s10875-021-00978-9
- Abhijit Anil Patil AD. Wiskott Aldrich syndrome having atypical presentation like Evans syndrome. Clin Pediatr (Phila). 2019;4:147.
- Kumar A, Jain S, Kumar P, Goyal JP. Generalised eczema: a diagnostic clue to Wiskott-Aldrich syndrome. BMJ Case Rep. 2021;14:242642. doi:10.1136/bcr-2021-242642
- Albert MH, Notarangelo LD, Ochs HD. Clinical spectrum, pathophysiology and treatment of the Wiskott-Aldrich syndrome. Curr Opin Hematol. 2011;18(1):42–48. doi:10.1097/MOH.0b013e32834114bc
- Thrasher AJ. WASp in immune-system organization and function. Nat Rev Immunol. 2002;2(9):635–646. doi:10.1038/nri884
- Villa A, Notarangelo L, Macchi P, et al. X-linked thrombocytopenia and Wiskott-Aldrich syndrome are allelic diseases with mutations in the WASP gene. Nat Genet. 1995;9(4):414–417. doi:10.1038/ng0495-414
- Schindelhauer D, Weiss M, Hellebrand H, et al. Wiskott-Aldrich syndrome: no strict genotype-phenotype correlations but clustering of missense mutations in the amino-terminal part of the WASP gene product. Hum Genet. 1996;98(1):68–76. doi:10.1007/s004390050162
- Worth AJ, Thrasher AJ. Current and emerging treatment options for Wiskott-Aldrich syndrome. Expert Rev Clin Immunol. 2015;11(9):1015–1032. doi:10.1586/1744666X.2015.1062366
- Suri D, Singh S, Rawat A, et al. Clinical profile and genetic basis of Wiskott-Aldrich syndrome at Chandigarh, North India. Asian Pacific J Allerg Immunol. 2012;30(1):71–78.
- Haskoloğlu Ş, Öztürk A, Öztürk G, et al. Clinical features and outcomes of 23 patients with Wiskott-Aldrich syndrome: a single-center experience. Turk J Hematol. 2020;37(4):271–281. doi:10.4274/tjh.galenos.2020.2020.0334
- Mullen CA, Anderson KD, Blaese RM. Splenectomy and/or bone marrow transplantation in management of the Wiskott-Aldrich syndrome: long term follow-up of 62 cases. Blood. 1993;82(10):2961–2966. doi:10.1182/blood.v82.10.2961.2961
- Ozsahin H, Cavazzana-Calvo M, Notarangelo LD. Long-term outcome following hematopoietic stem-cell transplantation in Wiskott-Aldrich syndrome: collaborative study of the European Society for Immunodeficiencies and European Group for Blood and Marrow Transplantation. Blood. 2008;111(1):439–445. doi:10.1182/blood-2007-03-076679
- Moratto D, Giliani S, Bonfim C, et al. Long-term outcome and lineage-specific chimerism in 194 patients with Wiskott-Aldrich syndrome treated by hematopoietic cell transplantation in the period 1980–2009: an international collaborative study. Blood. 2011;118(6):1675–1684. doi:10.1182/blood-2010-11-319376
- Jyonouchi S, Gwafila B, Gwalani LA, et al. Phase I trial of low-dose interleukin 2 therapy in patients with Wiskott-Aldrich syndrome. Clin Immunol. 2017;179:47–53. doi:10.1016/J.CLIM.2017.02.001
- Zaninetti C, Gresele P, Bertomoro A, et al. Eltrombopag for the treatment of inherited thrombocytopenias: a phase II clinical trial. Haematologica. 2020;105(3):820–828. doi:10.3324/haematol.2019.223966
- Gabelli M, Marzollo A, Notarangelo LD, Basso G, Putti MC. Eltrombopag use in a patient with Wiskott–Aldrich syndrome. Pediatr Blood Cancer. 2017;64(12):1–3. doi:10.1002/pbc.26692
- Gerrits AJ, Leven EA, Frelinger AL, et al. Effects of eltrombopag on platelet count and platelet activation in Wiskott-Aldrich syndrome/X-linked thrombocytopenia. Blood. 2015;126(11):1367–1378. doi:10.1182/blood-2014-09-602573
- Bach FH, Albertini RJ, Joo P, Anderson JL, Bortin MM. Bone-marrow transplantation in a patient with the Wiskott-Aldrich syndrome. Lancet. 1968;2(7583):1364–1366. doi:10.1016/s0140-6736(68)92672-x
- Filipovich AH, Stone JV, Tomany SC, et al. Impact of donor type on outcome of bone marrow transplantation for Wiskott-Aldrich syndrome: collaborative study of the International Bone Marrow Transplant Registry and the National Marrow Donor Program. Blood. 2001;97(6):1598–1603. doi:10.1182/blood.V97.6.1598
- Kobayashi R, Ariga T, Nonoyama S, et al. Outcome in patients with Wiskott–Aldrich syndrome following stem cell transplantation: an analysis of 57 patients in Japan. Br J Hematol. 2006;135:362–366. doi:10.1111/j.1365-2141.2006.06297.x
- Pai SY, Notarangelo LD. Hematopoietic cell transplantation for Wiskott-Aldrich syndrome: advances in biology and future directions for treatment. Immunol Allergy Clin North Am. 2010;30(2):179–194. doi:10.1016/j.iac.2010.02.001
- Shekhovtsova Z, Bonfim C, Ruggeri A, et al. A risk factor analysis of outcomes after unrelated cord blood transplantation for children with Wiskott-Aldrich syndrome. Haematologica. 2017;102(6):1112–1119. doi:10.3324/haematol.2016.158808
- Elfeky RA, Furtado-Silva JM, Chiesa R, et al. One hundred percent survival after transplantation of 34 patients with Wiskott-Aldrich syndrome over 20 years. J Allergy Clin Immunol. 2018;142(5):1654–1656.e7. doi:10.1016/j.jaci.2018.06.042
- Balashov D, Laberko A, Shcherbina A, et al. A conditioning regimen with plerixafor is safe and improves the outcome of TCRαβ + and CD19 + cell-depleted stem cell transplantation in patients with Wiskott-Aldrich syndrome. Biol Blood Marrow Transplant. 2018;24(7):1432–1440. doi:10.1016/j.bbmt.2018.03.006
- Albert MH, Bittner TC, Nonoyama S, et al. X-linked thrombocytopenia (XLT) due to WAS mutations: clinical characteristics, long-term outcome, and treatment options. Blood. 2010;115(16):3231–3238. doi:10.1182/blood-2009-09-239087
- Gennery AR, Slatter MA, Grandin L, et al. Transplantation of hematopoietic stem cells and long-term survival for primary immunodeficiencies in Europe: entering a new century, do we do better? J Allergy Clin Immunol. 2010;126(3):602–610.e11. doi:10.1016/j.jaci.2010.06.015
- Gennery AR, Cant AJ. The immunocompromised host: the patient with recurrent infection. Adv Exp Med Biol. 2004;549:109–117. doi:10.1007/978-1-4419-8993-2_16
- Seger RA, Gungor T, Belohradsky BH, et al. Treatment of chronic granulomatous disease with myeloablative conditioning and an unmodified hemopoietic allograft: a survey of the European experience, 1985–2000. Blood. 2002;100(13):4344–4350. doi:10.1182/blood-2002-02-0583
- Yue Y, Shi X, Song Z, et al. Posttransplant cyclophosphamide for haploidentical stem cell transplantation in children with Wiskott–Aldrich syndrome. Pediatr Blood Cancer. 2018;65(8):1–5. doi:10.1002/pbc.27092
- Mallhi KK, Petrovic A, Ochs HD. Hematopoietic stem cell therapy for Wiskott– Aldrich syndrome: improved outcome and quality of life. J Blood Med. 2021;12:435–447. doi:10.2147/JBM.S232650
- Yamaguchi K, Ariga T, Yamada M, et al. Mixed chimera status of 12 patients with Wiskott-Aldrich syndrome (WAS) after hematopoietic stem cell transplantation: evaluation by flow cytometric analysis of intracellular WAS protein expression. Blood. 2002;100(4):1208–1214. doi:10.1182/blood-2002-01-0211
- Slatter MA, Rao K, Abd Hamid IJ, et al. Treosulfan and fludarabine conditioning for hematopoietic stem cell transplantation in children with primary immunodeficiency: UK experience. Biol Blood Marrow Transplant. 2018;24(3):529–536. doi:10.1016/j.bbmt.2017.11.009
- Burroughs LM, Storb R, Leisenring WM, et al. Intensive postgrafting immune suppression combined with nonmyeloablative conditioning for transplantation of HLA-identical hematopoietic cell grafts: results of a pilot study for treatment of primary immunodeficiency disorders. Bone Marrow Transplant. 2007;40(7):633–642. doi:10.1038/sj.bmt.1705778
- Ngwube A, Hanson IC, Orange J, et al. Outcomes after allogeneic transplant in patients with Wiskott-Aldrich syndrome. Biol Blood Marrow Transplant. 2018;24(3):537–541. doi:10.1016/j.bbmt.2017.11.019
- Stepensky P, Krauss A, Goldstein G, et al. Impact of conditioning on outcome of hematopoietic stem cell transplantation for Wiskott-Aldrich syndrome. J Pediatr Hematol Oncol. 2013;35(6):234–238. doi:10.1097/MPH.0b013e318279cbfc
- Kamani NR, Kumar S, Hassebroek A, et al. Malignancies after hematopoietic cell transplantation for primary immune deficiencies: a report from the center for international blood and marrow transplant research. Biol Blood Marrow Transplant. 2011;17(12):1783–1789. doi:10.1016/j.bbmt.2011.05.008
- Curtis RE, Travis LB, Rowlings PA, et al. Risk of lymphoproliferative disorders after bone marrow transplantation: a multi-institutional study. Blood. 1999;94(7):2208–2216. doi:10.1182/blood.V94.7.2208.419k21_2208_2216
- Bulaklak K, Gersbach CA. The once and future gene therapy. Nat Commun. 2020;11(1):11–14. doi:10.1038/s41467-020-19505-2
- Braun CJ, Boztug K, Paruzynski A, et al. Gene therapy for Wiskott-Aldrich syndrome-long - term efficacy and genotoxicity (Science Translational Medicine). Sci Transl Med. 2014;6(254):1–15. doi:10.1126/scitranslmed.3010603
- Boztug K, Schmidt M, Schwarzer A, et al. Stem-cell gene therapy for the Wiskott–Aldrich syndrome. N Engl J Med. 2010;363(20):1918–1927. doi:10.1056/nejmoa1003548
- Hacein-Bey Abina S, Gaspar HB, Blondeau J, et al. Outcomes following gene therapy in patients with severe Wiskott-Aldrich syndrome. JAMA. 2015;313(15):1550–1563. doi:10.1001/jama.2015.3253
- Ferrua F, Cicalese MP, Galimberti S, et al. Lentiviral haemopoietic stem/progenitor cell gene therapy for treatment of Wiskott-Aldrich syndrome: interim results of a non-randomised, open-label, phase 1/2 clinical study. Lancet Haematol. 2019;6(5):e239–e253. doi:10.1016/S2352-3026(19)30021-3
- Morris EC, Fox T, Chakraverty R, et al. Gene therapy for Wiskott-Aldrich syndrome in a severely affected adult. Blood. 2017;130(11):1327–1335. doi:10.1182/blood-2017-04-777136
- Sereni L, Castiello MC, Di Silvestre D, et al. Lentiviral gene therapy corrects platelet phenotype and function in patients with Wiskott-Aldrich syndrome. J Allergy Clin Immunol. 2019;144(3):825–838. doi:10.1016/j.jaci.2019.03.012