
Abstract
Disorders of copper homeostasis are currently recognized across the life span. Their recognition and links to human disease have spanned several decades, beginning with the recognition of a degenerative disorder in the offspring of sheep grazing in copper-deficient pastures, through to the description of infants suffering from a progressive neurodegenerative disorder characterized by epileptic seizures, developmental regression, failure to thrive, and an unusual hair quality (giving the condition its distinctive label of “kinky hair disease”). In this review, we trace the historical background and describe the biochemistry and physiology of copper metabolism and transport, inheritance patterns, molecular genetics, and genotype–phenotype correlations based on current understanding of the disorder. It is clear from the clinical presentations and variants that disorders of copper homeostasis include phenotypes ranging from mild occipital horn syndrome to intermediate and severe forms of classical Menkes disease. The symptoms involve multiple organ systems such as brain, lung, gastrointestinal tract, urinary tract, connective tissue, and skin. A multisystem disorder needs a multidisciplinary approach to care, as treatment interventions permit longer survival for some individuals. Animal models have been developed to help screen treatment options and provide a better understanding of these disorders in the laboratory. Finally, we propose a multidisciplinary approach to promote continued research (both basic and clinical) to improve survival, quality of life, and care for these conditions.
Introduction and historical background
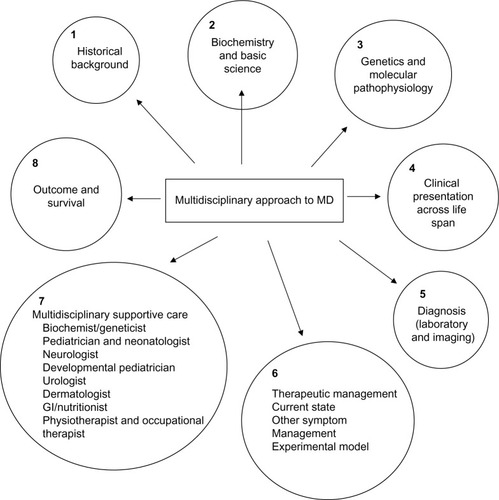
The focus of this review is to highlight the role a multidisciplinary approach can play in optimizing care of this multisystem lethal disease (). The history of Menkes disease (MD) dates back to as early as 1937 when Australian veterinary scientists recognized an association between copper deficiency and a demyelinating disease of the brain in the offspring of sheep grazing in copper-deficient pastures. Researchers in the 1960s observing defective wool in affected sheep turned to the Australian Wool Research Laboratories to establish a link between a copper-deficient diet and defective hair formation. However, efforts to establish a causal relationship between a copper-deficient diet and wool quality remained inconclusive.
Figure 1 Multidisciplinary approach to MD.
In 1962, Menkes et al at Columbia University, New York, NY, USA, described a distinctive clinical syndrome comprising neurological degeneration in five affected male infants of English–Irish heritage who showed unusual hair quality and failure to thrive. These male infants appeared normal at birth through early infancy; subsequently, they developed epileptic seizures and regression of developmental milestones progressing to death between the age of 7 months and 3.5 years.Citation1
O’Brien and SampsonCitation2 coined the term “kinky hair disease” and performed biochemical studies on frozen brain tissues of two siblings, which demonstrated a reduction in docosahexaenoic acid, the most highly unsaturated fatty acid in brain. Disruption of mitochondrial function was postulated as playing a possible role in this disorder on account of its involvement in fatty acid oxidation.
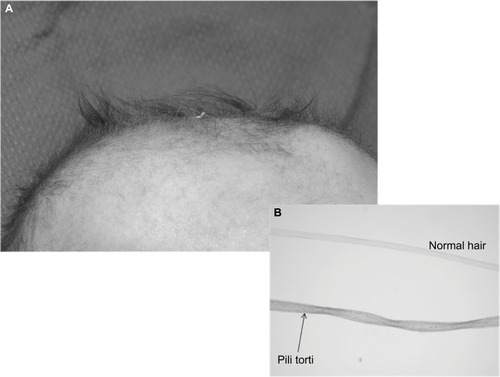
Danks et alCitation3 reported seven new cases of Menkes kinky hair syndrome from five families seen over a period of 3 years in Melbourne, Australia. The mode of inheritance in each pedigree was compatible with X-linked Mendelian pattern. Hypothermia, developmental delay, growth failure, and very low serum copper were noted along with the unusual hair quality of infants. Hairs from the scalp in these infants appeared sparse, tangled, and lusterless with rotational twists of “pili torti”, which were similar in texture to the brittle wool of sheep raised on copper-deficient soil in Australia (). Sections of the scalp showed hair follicles deeply buried in the dermis with small, brittle, and broken hair shafts.
Figure 2 MD: (A) scalp shows “kinky hair”, (B) the inset shows “pili torti” and a normal hair strand under a high power microscope.
MD, as it is currently known, is a lethal X-linked disorder of copper metabolism with multisystem involvement. This disease shows wide variability and clinical heterogeneity in manifestations, ranging from a severe clinical course leading to death in early childhood to a milder form termed occipital horn syndrome (OHS) with connective tissue abnormalities and longer survival.Citation4 Patients with MD with intermediate phenotypes (5%–10%) characterized by later onset and milder symptoms are also described.Citation4,Citation5 These variant presentations are being better understood with evolving genotype–phenotype correlations.
Danks et alCitation6 who carried out further research on this disorder used another term, “steely hair disease”. A number of other eponyms, such as Menkes steely hair disease, Menkes kinky hair disease, trichopoliodystrophy, and Menkes syndrome, have been used in scientific nomenclature.
It is important to see how current understanding of different aspects of this disorder has been shaped by developments in the fields of biochemistry, molecular genetics, pathology, clinical presentation, and therapeutic interventions.
Epidemiology
The overall incidence of MD is reported to be 1 in 100,000–250,000 births.Citation7 However, regional geographic variation is recognized. The incidence in Australia is reported to be much higher than elsewhere at 1:50,000–100,000.Citation5 A large observational study reported the incidence as 1:300,000 in Europe and 1:360,000 in Japan.Citation7,Citation8 The true incidence rate of OHS is not reported in the literature (to the authors’ knowledge), but it is believed to be lower than that of MD. Approximately 35–40 affected individuals are reported in the literature, with the possibility that the condition may be underdiagnosed.Citation9,Citation10
The vast majority of patients are male, while biological mothers are heterozygotes or carriers. The population frequency of sporadic cases reported is compatible with the one-third expected for an X-linked lethal disease with equal mutation rates in egg and sperm, which implies a mutation rate of 6.7×10−6/gamete/generation. However, the literature reports few affected female patients, with most of them having an X-autosome translocation with a breakpoint at Xq21.1, truncating ATPA7, which results in clinical manifestations and is attributed to random X inactivation.Citation4,Citation11 In addition, there are few reports of females with milder symptoms who carry exon deletions (exon 6, 6–9, 1) or base-pair substitutions.Citation12–Citation14
Biochemistry
The liver and kidney are the two organs in mammals that have the highest concentration of copper.Citation15 A healthy adult body contains ~110 mg of copper, which is distributed in the liver, kidney, brain, blood, and skeletal tissues.Citation16 Vegetables, animal proteins, and human breast milk are good sources of copper.Citation17,Citation18 Under normal dietary conditions, the gastrointestinal (GI) tract in humans absorbs ~30%–40% of ingested copper.Citation18 Copper is present in various GI fluids such as salivary, gastric, pancreatic, duodenal, and bile. The absorption of copper in humans occurs in two phases.Citation15 In the first phase, copper is absorbed from the distal small intestine and reabsorbed into the circulation in its protein-bound form to reach the liver and kidney. Once copper enters the body, it is transported to different organs bound to plasma protein carriers (ceruloplasmin, transcuprein, and albumin).Citation19,Citation20 In the second phase, copper is transported from the liver (and perhaps kidney) to other organs, including the brain, heart, and skeletal tissue. A small amount of copper is secreted into the bile and is lost via the feces. Most of the copper reaching the liver and kidney is returned to plasma bound to ceruloplasmin, indicating that both organs may be important sources of protein-bound forms of copper in circulation.Citation16
Copper is an essential trace element that exists in two different oxidation states, Cu+ and Cu++. These two states play a crucial role in several copper enzyme-dependent metabolic processes. On the one hand, the capability of being able to switch oxidation states is important in metabolic processes, but on the other the same property can result in the release of free radicals adversely impacting cellular events. Therefore, maintenance of cellular homeostasis of copper is vital.
There are various enzymes that require copper for important biological activities within the human body (). These are cytochrome c oxidase (COX) for cellular respiration; dopamine β-hydroxylase for biosynthesis of neurotransmitters; lysyl and sulfhydryl oxidase for collagen, elastin, and keratin cross-linking; tyrosinase for pigmentation; and superoxide dismutase for free-radical scavenging. Ceruloplasmin, a key circulating copper-binding protein has a crucial role in the regulation of copper and iron homeostasis.Citation5,Citation21 The impact of copper deficiency in the nervous system is significant to the current understanding of neuronal dysfunction at a cellular level and its effects on neuropathogenesis of MD. Low levels of brain copper in MD affect catecholamine pathways, leading to abnormal catecholamine metabolite levels in blood and body fluids. While norepinephrine levels were found to be normal in the infants studied, the ratios of the dopamine metabolites, dihydroxyphenylacetic acid (DOPAC) and the catecholamine precursor dihydroxyphenylalanine, to neuronal metabolite of norepinephrine, dihydroxyphenylglycol (DHPG), were increased above control values, suggesting partial dopamine β-hydroxylase deficiency. This forms the basis of a newborn screening test, which is discussed in the following sections. Furthermore, a significant reduction in brain COX activity leads to intracerebral lactic acidosis and other mitochondrial abnormalities in MD, which points to insufficiency in cellular bioenergetics as a contributor to neuronal dysfunction.
Table 1 Copper enzymes and their role in MD
Gene expression profiling in the single postmortem study of a patient with MD identified dysregulation of 394 genes in the cerebellum and 121 genes in the cerebral cortex. At least 30 genes were found to be common to both the cortex and cerebellum. These genes encode enzymes in the tricarboxylic acid cycle and oxidative phosphorylation (mitochondrial adenosine triphosphate [ATP] synthase), as well as affect ribosomal translation mechanisms, signal transduction, and immune responses. It has also been suggested that the ATP7A protein plays a role in the modulation of excitotoxicity. In vitro studies of mouse models of MD indicate that atp7a-mediated release of copper from an intracellular pool in response to calcium-mediated N-methyl-d-aspartate receptor activation is impaired, leaving the neuron vulnerable to excitotoxic injury.
Therefore, in MD, a copper-deficient state is characterized by a multisystem involvement that reflects biochemical perturbation mediated through copper deficiency states that affect enzymatic function involving critical biological processes. The resulting deficits in neurotransmitter function, energy metabolism, and excitotoxicity play key roles in the neurological manifestations of the disease.
Copper metabolism in the cell
Copper uptake across plasma membrane is dependent on membrane transporter (CTR1).Citation22 Copper is bound to various proteins such as metallothionein, glutathione, or copper-specific chaperones that have a role in copper loading of enzymes.Citation23,Citation24 Three types of copper chaperones are known: ATOX1, COX 17, and CCS. ATOX1 helps copper to reach the trans-Golgi network, where it is incorporated into two copper-specific ATPases: ATP7A and ATP7B.Citation25 Dysfunction of these two ATPases causes MD and Wilson disease, respectively. ATP7A is expressed in almost every organ except liver, while ATP7B is expressed predominantly in the liver. This explains why MD is a systemic disease and Wilson disease is primarily a liver-specific disease with some neurological and ophthalmological involvement.
COX 17 directs the copper into mitochondria where other copper metalation proteins such as COX 11, SCO1, and SCO2 are present,Citation26 while CCS directs copper to superoxide dismutase, which resides in the cytosol or the mitochondria.Citation27 ATP7A and ATP7B have important roles in ATP-driven cellular efflux of copper as described in the following section.Citation28
Regulation of copper transport
The ATP7A mRNA is expressed in several human tissues, but is notably low or absent in the liver, while the ATP7A gene product is a transmembrane protein localized to the Golgi network. Under normal physiological conditions where the copper concentration in the body is normal, ATP7A remains confined within the trans-Golgi network and continues to transport copper to the copper-dependent enzymes. However, when total intracellular copper stores increase, ATP7A relocates to the cell membrane to promote copper efflux.Citation29,Citation30 At the blood–brain barrier, ATP7A tends to traffic to the basolateral membrane promoting delivery of copper from blood to the brain, while in certain specialized epithelial cells such as the choroid plexus, the protein localizes to the apical membrane. Once copper reaches the cerebrospinal fluid, it forms complexes with albumin and amino acids (histidine). It is then imported into neurons through specific copper importers (DMT1 and CRT1) and delivered through metallochaperones to ATP7A and ATP7B, eventually participating in the metalation of cuproenzymes.
ATP7A is a multitasking protein that is involved in a multitude of functions ranging from axonal outgrowth, synaptic integrity, responses to N-methyl-d-aspartate receptor activation, and other functions as listed in .
Table 2 Functions of ATP7A: a multitasking protein in the nervous system
Molecular genetics
Early studies in understanding the genetics of MD came through the identification of a defect in copper metabolism.Citation39 Copper concentrations were elevated in the cultured fibroblasts of affected males and heterozygote carriers. Similarities between the clinical and biochemical phenotype in humans and the “mottled” mouse (Mo) and the knowledge that mottled gene mutations were close to the phosphoglycerate kinase (PGK) gene, which in humans is located on the X chromosome, led to the conclusion that the gene for MD was likely located on the X chromosome. Further, linkage studies using restriction fragment length polymorphism markers in a large kindred and studies on a female patient with both this disease and an X- autosome translocation with karyotype 46, X, t(X; 2) (q13; q32.2) indicated that the Menkes gene maps to a small subregion of band Xq13.2-q13.3 proximal to the PGK1 locus,Citation40 which was also confirmed by additional fine-mapping studies.Citation41 A yeast artificial chromosome clone was isolated spanning the breakpoint region, and cDNA libraries were scanned using phage subclones from the breakpoint region. An 8 kb transcript from a gene at the breakpoint showed diminished hybridization in patients with MD. Partial sequencing identified metal-binding motifs supporting the likelihood that this was indeed the candidate gene. The ATP7A gene is located at the chromosome locus Xq13.3 and has eight transmembrane segments and six copper-binding sites/domains, additional domains for phosphatase, phosphorylation, transduction, and ATP binding.
Using restriction enzyme mapping, exon identification, and sequencing techniques, 23 exons were identified spanning a 140 kb region as well as the intron–exon boundaries defining the ATP7A gene. The gene was found to bear a strong sequence homology at the 3′ region with the Wilson disease (Wnd) gene and divergence for a third of the gene at the 5′ region, suggesting a common ancestral origin from an evolutionary perspective. Further characterization of the candidate gene for Menkes (MNK) disclosed that the gene codes for a 1,500 amino-acid protein, which is predicted to be a P-type cation-transporting ATPase (ATP7A) similar to a bacterial copper-transporting ATPase.Citation42–Citation44
Most of the clinical phenotypic features were already attributable to malfunction of copper-containing enzymes that were deficient in copper, and cell studies from patients with MD had already been demonstrated to accumulate increased amounts of copper attributable to a reduced efflux of the copper ion. These findings were in line with the discovery of the candidate gene product bearing a homology to heavy metal-binding ATPases in bacteria and man. With these discoveries, MD has come to be understood as a disorder of copper homeostasis.
Genotype–phenotype correlations
Pathogenic mutations in ATP7A result in a variety of clinically recognizable phenotypes associated with copper transport disorders (MD, OHS, and the ATP7A-related distal motor neuropathy). The degree to which residual ATP7A function is maintained determines the severity of the clinical phenotypes. Mutations of every variety have been associated with MD; the majority are de novo and private. Exon deletions can be detected by the multiplex ligation-dependent probe amplification as an initial DNA test, while smaller intragenic mutations can be examined by polymerase chain reaction amplification and sequencing of coding exons using genomic DNA.
More than 311 different mutations have been reported in patients with MD.Citation45 Deletions or insertions (22%), large gene deletions (22%), duplications, missense (17%), nonsense (18%), and splice junction mutations (18%) have all been described. A 700-nucleotide region involving exons 7–10 seem to be associated with a significant proportion of pathogenic mutations resulting in MD.Citation46–Citation48 Those mutations that result in a severe truncation of the gene product and significant loss of function likely result in MD; hypofunctional splice site resulting from aberrant splicing and missense mutations with retention of protein function are associated with milder phenotypes. With 0%–15% residual function in ATP7A activity, the resulting phenotype is MD; higher degrees of function up to 30% result in the milder OHS phenotype, while 60%–70% residual function is associated with late-onset distal motor neuropathy.Citation49
Although the majority of affected individuals are males, heterozygous females tend to be carriers based on skewed inactivation of the X chromosome; rarely females with chromosomal translocations have been reported to be affected and manifest symptoms.
Of all the different mutations described,Citation50 two mutations resulting in MD are amenable to early treatment with copper (Gly666Arg and Gly727Arg). Not much is known about the treatment response of remaining missense mutations.
The OHS is an allelic disorder of MD that results from exon skipping and aberrant splicing, although missense mutations without aberrant splicing are also described. Less frequently, deletions affecting the promoter region or resulting in a leaky “splice junction” may also lead to the milder OHS phenotype. The mutations seem to result in a significantly greater degree of retention of ATP7A activity than those leading to MD.
The third phenotype of ATP7A-related distal motor neuropathy is distinctive in that there are no overt copper-related metabolic/biochemical abnormalities. The mutations underlying this phenotype are missense mutations affecting amino acids within or near transmembrane segments of the protein. The resulting amino acid substitutions appear to affect trafficking of the protein to distal axons and the resulting deficiency of copper-dependent enzymes critical to the maintenance of integrity and function of the motor neuron and its axons. The precise mechanism underlying the pathophysiology of this unique disorder of copper transport and metabolism remains to be elucidated.Citation51–Citation54
Clinical presentation of MD
The key clinical features of classical MD, intermediate forms, and OHS are summarized in .
Table 3 Manifestations of MD
Classical form of MD
Pregnancy and labor are usually uncomplicated, and most affected males born with MD have normal anthropometric measures at birth. However, early features noted with these neonates are: hypothermia, cephalohematomas, persistent hyperbilirubinemia, hypoglycemia, and feeding difficulties.Citation30,Citation55,Citation56 Such infants are also recognized to carry subtle dysmorphic facial features such as frontal or occipital prominence, pudgy cheeks with broad nasal bridge, and micrognathia and often may appear dull and expressionless.Citation55 Various types of congenital malformations are known to be associated with MD. In one study, 35 patients with MD were reviewed and 14 were found to have congenital malformations. Out of these 14 patients, eight had minor congenital malformations including high arched palate, micrognathia, congenital microblepharia, entropion, and a flat occiput. The remainder had major congenital malformations including cerebellar hypoplasia, cystic white-matter changes in the brain, congenital complete A–V block, and cystic changes in the arachnoid membranes in the lungs.Citation57
Respiratory infections such as pneumonias are reported in the literature as one of the causes of severe morbidity or mortality in MD.Citation58,Citation59 An anecdotal report of a 19-month-old male infant who died of MD revealed, at necropsy, bilateral diffuse panlobular emphysema in the lungs.Citation60 A similar case is also reported in an infant presenting with respiratory illness from the age of 9 months, in whom chest radiography revealed progressive generalized hyperinflation and increased cystic spaces. An autopsy at 14 months showed severe diffuse panlobular emphysema with large bullae along with abnormal pulmonary vascular development.Citation56 The authors hypothesized that deficiency of the copper-dependent enzyme, lysyl oxidase, leads to abnormalities in quality and quantity of elastin, which forms an important element in connective tissue formation, leading to aneurysmal dilatation and emphysema in MD.
GI manifestations in MD are reported infrequently, and there may be an under-recognition of involvement of the GI tract. Gastric polyps, gastroesophageal reflux disease, and colonic diverticula are reported in the literature.Citation56,Citation61,Citation62 Hiatus hernia has been reported to be associated with MD and OHS.Citation63,Citation64 Progressive sliding hiatus hernia was noted in a 19-month-old male infant with MD, which progressed rapidly, requiring surgical intervention.Citation63 An additional two infants with MD have been described in whom severe GI bleeding secondary to gastric polyps was noted. One of these patients died of severe hemorrhage.Citation65 In addition to GI manifestations, an unusual presentation of MD with hepatomegaly in a 4-month-old infant who was known to carry a missense mutation of his ATP7A gene (Gly727Arg mutation) has also been reported. The liver biopsy revealed nonspecific cholestasis and focal necrosis.Citation66
Several connective tissue abnormalities are described in MD, which are considered to be secondary to reduced activity of lysyl oxidase, a copper-dependent enzyme important for cross-linking collagen and elastin.Citation67,Citation68 These include pectus excavatum, pectus carinatum, the presence of wormian bones in skull, osteopenia, and spontaneous fractures of long bone.Citation59 Most patients with MD have mottled hypopigmented and pale skin presumably due to tyrosinase deficiency.Citation69
The scalp hairs () may appear normal at birth, but by 2–3 months of age they become kinky, coarse, sparse, brittle, and hypopigmented, which are characteristic of MD.Citation70,Citation71 Microscopic examination of the hair reveals various abnormalities and even aids in the diagnosis of milder form of the disease.Citation72 Several hair shaft abnormalities have been reported, with pili torti (twisted hair) being the most common; trichorrhexis nodosa (fracture of the hair shaft at regular intervals), monilethrix (varying diameter of hair shafts), trichoclasis (hairs are broken off from 6 mm to 8 mm from the skin), and trichoptilosis (splitting of the shaft of the hair) have also been reported.Citation72,Citation73
The neurological manifestations of MD are also varied. Epileptic seizures are one of the most common and well-recognized features of MD. Most patients develop treatment-resistant forms of seizures by ~2–3 months of age.Citation74,Citation75 Many different kinds of seizures have been described in affected infants and children with MD. Seizures in MD can be triggered by the acute stress secondary to severe infection, a febrile event, or could be preceded by nonconvulsive seizures associated with recurrent episodes of apnea.Citation76–Citation79 A case series (Bahi-Buisson et al) focused on electroclinical features in MD and described the natural progression of epilepsy in MD; the key features are summarized in .
Table 4 Evolution and progression of epilepsy in MD
Seizures and neurological regression usually become apparent at 6–8 weeks of life. Three stages are identified in the natural history of the progression of epilepsy in MD. Electroencephalography (EEG) abnormalities may antedate the appearance of seizures, as early as 2 months of life. The three stages are as follows: 1) an early stage at the age of 3 months with focal seizures progressing to status epilepticus, 2) an intermediate stage (6–11 months) where epileptic spasms, including West syndrome accompanied by EEG changes of hypsarrhythmia, develop; myoclonic seizures as a presenting symptom have also been reported, and finally, 3) a third late stage that is characterized by the emergence of multifocal seizures, tonic spasms, and myoclonus encountered in infants with a mean age of ~20–25 months.Citation78,Citation80,Citation81 Typical interictal and ictal EEG findings are described in an earlier articleCitation78,Citation81 ().
Figure 3 EEG segments from patient with MD.
Abbreviations: EEG, electroencephalography; MD, Menkes disease.
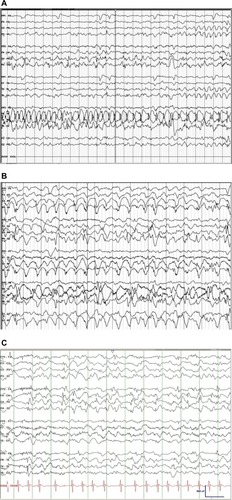
EEG abnormalities correlate with molecular defects, as shown in a series of 24 patients with MD, where patients with missense and leaky splice junction mutations were found to have normal EEGs. On the other hand, large deletions, small deletions, nonsense mutations, and canonical splice junction mutations were associated with significant EEG abnormalities in >50% of the cases.Citation47,Citation74
Presymptomatic diagnosis and early treatment may improve brain electrical activity. This was demonstrated in a study which looked at 24 patients with MD. In that study, the authors reported that patients were started on copper supplementation soon after the diagnosis (≤6 weeks of age) and they noted clinical seizure in only 12.5% of the patients, and they found that 46% had at least one abnormal EEG tracing.Citation81 Early diagnosis and treatment are closely correlated with the genesis of epilepsy in MD. A study in 2008 demonstrated that copper replacement within 22 days of life in 12 newborns diagnosed with MD had improved the clinical outcome.Citation82
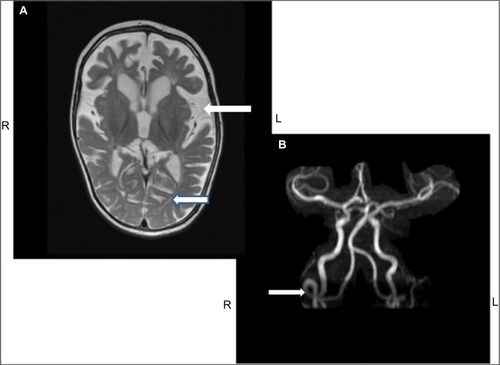
Neuropathologic changes in MD are also distinct. There is diffuse cerebral and cerebellar atrophy that is a consequence of volume loss of both white and gray matter. Widespread histological abnormalities noted include neuronal loss and gliosis within cortical gray matter, cystic infarction in the basal ganglia, loss of Purkinje cells in cerebellum, and spongy changes in cerebral white matter along with loss of myelin sheaths and axons.Citation83,Citation84 The muscle shows ragged red fibers on histology, with pleomorphism of mitochondria and cristae under the electron microscope.Citation82 Vascular abnormalities are noted in the cerebral arteries; these vessels appear thin walled, ectatic, and tortuousCitation59,Citation84 ().
Figure 4 MRI of the brain (A) and MRA (B) from the same patient.
Abbreviations: L, left; MRA, magnetic resonance angiography; MRI, magnetic resonance imaging; R, right.
Occipital horn syndrome
OHS is considered to be the mildest form of MD with a few male patients with overlapping features of classical and/or mild MD and OHS reported.Citation85 Patients with OHS have very similar presentations as with MD at birth, such as hypothermia, jaundice, and feeding difficulties. In addition, they may present with umbilical and inguinal hernias. The findings in contrast to classical MD are connective tissue abnormalities such as, wrinkled, loose skin and bony abnormalities of flat and long bones, including osteoporosis. The distinct radiologic feature of OHS is symmetric exostoses protruding from occipital bones, leading to the designation of “occipital horn”. The diagnosis of OHS is often delayed until 5–10 years of age due to milder and subtle symptoms. Usually, the early signs that bring these patients to medical attention are intractable diarrhea and recurrent urinary tract infection, which are secondary to bladder diverticula. Intellectual capabilities in these patients are described as low to borderline normal. Motor development is also delayed secondary to muscular hypotonia and other connective tissue disorders. Patients with OHS can remain alive until adulthood.Citation5,Citation86
Intermediate forms
Between classical MD and OHS lies another group of disorders that is a milder form of MD with late presenting symptoms.Citation87 These patients have abnormal hair (pili torti), variable connective tissue manifestations such as lax skin and joints, and characteristic facial features. They are also reported to have milder neurological presentations compared to MD and commonly have ataxia, dysarthria, and mild intellectual disabilities.Citation88,Citation89
Diagnosis
Early diagnosis of MD is challenging because clinical features and biochemical markers may be nonspecific and unreliable. Clinical presentations such as hypothermia, hyperbilirubinemia, and feeding difficulties are nonspecific and may present in neonates with other conditions such as sepsis. Serum copper and ceruloplasmin levels may be low in normal healthy newborns and hence not a good predictor in the early diagnosis of MD. Molecular analysis confirms the diagnosis. There are >300 different mutations reported in MD, which include missense and nonsense mutations along with deletion and insertions of one or more base pairs leading to frameshift and splice site mutations.Citation90–Citation92
Newborn screening
Currently, there are no newborn screening programs that are routinely screening for MD in countries where newborn screening programs exist for other genetic/metabolic disorders. However, a role for newborn screening can be rationalized on the basis of the evidence that early diagnosis seems to make a difference in survival and outcome in some patients with MD.Citation82 Measurement of neurochemicals such as dopamine, norepinephrine, DOPAC, and DHPG in the plasma and cerebrospinal fluid is reported to be highly specific and sensitive test to diagnose MD in the early neonatal period. The ratio of DOPAC to DHPG reflects the deficiency of dopamine β-hydroxylase activity.Citation82,Citation93
An argument can thus be made for the inclusion of MD into neonatal screening programs based on the detection and measurement of plasma dopamine, norepinephrine, DOPAC, and DHPG using blood dot specimens.
Prenatal diagnosis
Prenatal diagnosis is available for women who are MD carriers, by chorionic villus sampling and analyzing copper uptake in cultured fibroblasts in pregnancies at high risk, and where the mutation is unknown. If there is an affected individual in the family with a known mutation, then the pathogenic mutation can be identified through mutation analysis.Citation94,Citation95
Animal models
Much of our understanding about MD stems from animal models.Citation96 Early studies have focused on the mottled mouse that carries mutations in the Atp7a gene resulting in a mottled coat in heterozygous females. The murine gene locus Atp7a and the human homolog ATP7A are located on the X chromosome, and share >80% homology. Both gene products on sequence alignment show highly conserved amino acid sequences within functional domains.Citation97 These similarities extend to the ATP7A proteins in zebrafish and fruitfly. The initial description of the mottled mouse appeared in 1953,Citation98 and the defects in copper transport were described in these mutant mice by Hunt in 1974.Citation99 Currently, 109 mottled alleles are known in the Mouse Genome Informatics database. These have arisen spontaneously or by chemical, radiation, or targeted mutagenesis. Phenotypic severity in these models involving male mice varies from lethality in utero and death in 3 weeks to mice that survive a few postnatal months. A detailed assessment of these mutants is beyond the scope of this review. The reader is referred to an outstanding review on the subject.Citation97 It is however important to summarize important insights that have been gained. Mutations that confer embryonic lethality completely abolish residual protein activity, suggesting a critical role for copper in embryogenesis. “Brindled”, “mosaic”, and “macular” are strains that have been studied, and mutations in this group are missense or in frame deletions that lead to reduced synthesis of the mutant protein with reduced residual activity. Hemizygous males show characteristic features of severe copper deficiency, but survive beyond 3 weeks of postnatal life. Mutants that include “viable brindled” and “blotchy” survive a few months and develop features of atypical MD and/or OHS particularly connective tissue abnormalities. Aortic aneurysms (viable brindled), emphysema, osteoarthrosis, abdominal aneurysms (mottled), infertility (common to both mutants), and hypopigmentation are among the features shared between the models and human disease.
These mice have also served as models for exploring treatments. Copper replacement therapy given early to mottled mice improves survival, and the combination therapy with a copper chelator diethyldithiocarbamate has shown promising results.Citation100–Citation102 Treated animals showed improved survival, increased copper levels in the brain, improved locomotion, and greater body mass.
Transgenic mice models have also been developed from the brindled strain, where human ATP7A was expressed in a truncated form using recombinant adenovirus encoding the truncated gene that was administered by intraventricular injections. The treated mice survived longer and demonstrated higher dopamine monooxygenase activity in the brain along with improved neuromuscular strength and balance.
Mottled mice have also proven invaluable in studying the interaction between copper and iron metabolism suggesting intricate linkages that impact hematopoiesis and red cell turnover.
Other animal models include the zebrafish (Danio rerio) and the fruitfly (Drosophila melanogaster). In the zebrafish, calamity mutants generated by chemical mutagenesis have served as novel vertebrate models of MD.Citation103 The phenotypic effects extend to lack of pigmentation and defects of the notochord. Morpholinos (oligonucleotides that can target RNA sequence of interest) can correct aberrant splicing in the embryonic calamity mutant zebrafish and effect a rescue by leading to the production of normal Atp7a protein as in the wild type.Citation104
The fruitfly has been studied as a model for many neurodegenerative disorders. A null mutation in the DmATP7 gene has resulted in a classical model for MD.Citation105 The mutant fruitfly is extremely lethargic, while larvae are small and hypopigmented. Another model using conditional silencing of DmATP7 only in the digestive tract results in enhanced lethality and 50% survival to adulthood. The survivors show small brain size, but normal morphology as far as pigmentation is concerned. They are also sensitized to oxidative stress, a feature of MD that has been attributed to reduced superoxide dismutase activity.Citation106
Animal models can thus be used to develop therapeutic approaches through genetic modification as well as screening of compounds that affect copper metabolism.
Management
MD is a fatal disorder with death usually occurring between 6 months and 3 years of age as a result of neurological deterioration, poor feeding, aspiration, or respiratory infections. Those patients with less severe disease have a better survival rate with copper supplementation; however, the outcome depends on the severity of the ATP7A mutation and the residual function of ATP7A protein. Treatment with copper supplementation is most likely effective in those cases where the mutated protein retains the ability to transport copper across the blood–brain barrier. In patients with MD where the mutation leads to severe disease, treatment is ineffective.Citation82 Although treatment has not yet been known to reverse the neurological damage, a small subgroup of patients with MD manifesting milder symptoms have been reported to have better outcome.Citation107
There have been few reports of long-term survival with MD. Tchan et alCitation108 reported a patient with MD at 34 years having received copper supplementation for >30 years and having only mild intellectual impairment. Another patient with MD receiving copper supplement was reported to have survived till the age of 13.5 years.Citation109
Parenteral copper administration is considered the mainstay in treatment protocols for MD as oral preparations are not shown to be effective. Most researchers have used copper histidinate for treating MD.Citation110,Citation111 Subcutaneous administration of copper histidinate can normalize circulating blood levels of copper and ceruloplasmin and may replete brain copper.Citation112
Prasad et alCitation113 reported the correction of biochemical markers in a patient with MD using subcutaneous cupric chloride. They reported restoration of serum copper and ceruloplasmin along with clinical improvement in skin color and quality of hair; however, neurological status did not improve.
Early screening and prompt medical intervention are crucial in MD. Patients who were diagnosed and received early treatment with copper (within 25 days) had a much better neurodevelopmental outcome compared to late onset of treatment (228 days).Citation114 KalerCitation112 conducted a clinical trial using copper histidinate for MD, looking at somatic growth and neurodevelopmental outcome in 60 patients. The findings in this trial suggested that those who received copper histidinate before the onset of symptoms showed significant improvements in gross motor, fine motor/adaptive, personal–social, and language development in comparison to those who were symptomatic at the time of diagnosis. The mortality rate at the age of 3 years was also reported to be higher in the symptomatic group.Citation112 Other studies have also reported similar outcomes.Citation115,Citation116 Christodoulou et alCitation111 reported improvement in muscular tone and seizure activities in four patients with MD who they followed as a result of early treatment with copper histidine for 10–20 years. However, the response to connective tissue abnormalities associated with MD was noted to be poor.
Multidisciplinary approach to MD
MD is a multisystem disorder that has no cure. Current published data on MD suggests that there is wider age dependent variability in phenotypes at presentation.Citation54 However, contrary to previous notions of shortened life expectancy, patients currently seem to have better outcomes in terms of survival. A multidisciplinary approach can improve the quality of life and holistic care for patients with MD ().
Table 5 Multidisciplinary management in MD
Neonatal nurses or neonatologists can play an important role in the early recognition of subtle signs and symptoms such as hypothermia, unusual hair, and dysmorphic facial features. This can facilitate early referral and diagnosis, supporting the initiation of copper supplementation as required. Outside the neonatal unit, nurses or doctors in clinics or in the emergency department can come across this disorder. Children may present with macrocephaly with subdural hematoma, occasionally mimicking a diagnosis of shaken baby syndrome, needing further evaluation.Citation117 Patients with MD may also present with macrocephaly and seizures closely simulating neurological disorders such as leukodystrophies and Canavan or Alexander diseasesCitation118,Citation119 prompting early referral to pediatric neurologists and genetic metabolic specialists. These patients may also present to developmental or neurology clinics with a history of delayed or regressing milestones.
Interventions involve the institution of copper replacement therapy and the potential use of chaperones in the future. Failure to thrive should be assessed by a feeding team in association with a dietitian who can provide a feeding plan to optimize growth. Occupational therapists and physiotherapists can provide detailed assessment and management strategies for those patients who have problems with their tone and mobility. These can include a range of movement exercises, ways to prevent contractures and bedsores as well as assisting in formulating plans for activities of daily living.
Parents of patients with MD can benefit from education on how to give injections and take care of the patients at home. Anticipatory guidance about creating a safe environment, medications to alleviate symptoms, and supportive interventions for seizure management can be provided through nurse educators.
Genetic counselors have a role in providing parental reproductive risk counseling and prenatal diagnosis. Parents benefit through the sharing of information through support groups and social media connectivity. It is often noted that families taking care of patients with very high needs face emotional, financial, and social challenges. A social worker can assist these families by providing counseling services and financial support and also help link families to support groups and the national organization of rare disorders.
Conclusion
The past several decades have witnessed rapid and tremendous advances in our understanding of MD and related disorders of copper homeostasis. This review highlights the advances and breakthroughs in the basic science and molecular genetics leading to improved recognition and diagnosis. Early diagnosis has been shown to improve survival, and the availability of copper replacement therapy is a step forward. Multidisciplinary research and care teams can not only help in the development of clinical trials on MD in the future but also further the goal of improving quality of life for survivors. The work in basic science through the development of animal models holds great promise for future therapies for this otherwise fatal disorder.
Acknowledgments
The authors would like to thank CA Rupar, PhD, Professor, Departments of Pathology and Laboratory Medicine, Biochemistry, and Pediatrics, for his critical review of the manuscript, comments, and suggestions. There was no funding source for this review.
Disclosure
Asuri N Prasad has received honoraria from EMD Serono Inc in the last financial year. The authors report no other conflicts of interest in this work.
References
- MenkesJHAlterMSteiglederGKWeakelyDRSungJHA sex-linked recessive disorder with retardation of growth, peculiar hair, and focal cerebral and cerebellar degenerationPediatrics19622976477914472668
- O’BrienJSSampsonELKinky hair disease. II. Biochemical studiesJ Neuropathol Exp Neurol19662545235305922551
- DanksDMCampbellPEStevensBJMayneVCartwrightEMenkes’s kinky hair syndrome; an inherited defect in copper absorption with widespread effectsPediatrics19725021882015045349
- TimerZHornNMenkes Disease: recent advances and new aspectsJ Med Genet19973442652749138147
- TumerZMollerLBMenkes DiseaseEur J Hum Genet201018551151819888294
- DanksDMCartwrightEStevensBJTownleyRRMenkes’ kinky hair disease: further definition of the defect in copper transportScience197317978114011424120259
- TønnesenTKleijerWJHornNIncidence of Menkes diseaseHum Genet19918644084101999344
- GuYHKodamaHShigaKNakataSYanagawaYOzawaHA survey of Japanese patients with MD from 1990 to 2003: incidence and early signs before typical symptomatic onset, pointing the way to earlier diagnosisJ Inherit Metab Dis200528447347815902550
- TsukaharaMImaizumiKKawaiSKajiiTOccipital horn syndrome: report of a patient and review of the literatureClin Genet199445132358149649
- HornNMortonNERaoDCGenetic epidemiology of Menkes diseaseGenet Epidemiol1986342252303744020
- KapurSHigginsJVDelpKRogersBMenkes syndrome in a girl with X-autosome translocationAm J Med Genet19872625035103812600
- SirletoPSuraceCSantosHLyonization effects of the t(X; 16) translocation on the phenotypic expression in a rare female with MDPediatr Res200965334735119092723
- DesaiVDonsanteASwobodaKJMartensenMThompsonJKalerSGFavorably skewed X-inactivation accounts for neurological sparing in female carriers of Menkes diseaseClin Genet201179217618220497190
- MollerLBLenartowiczMZabotMTClinical expression of Menkes disease in females with normal karyotypeOrphanet J Rare Dis20127622264391
- LinderMCThe Biochemistry of CopperNew York, NYPlenum Press199173134
- LinderMCWootenLCervezaPCottonSShulzeRLomeliNCopper transportAm J Clin Nutr1998675 supplS965S971
- VuoriEKuttunenPThe concentrations of copper and zinc in human milk. A longitudinal studyActa Paediatr Scand19796813337758729
- WapnirRACopper absorption and bioavailabilityAm J Clin Nutr1998675 Suppl10541060
- TurnlundJRKeyesWRAndersonHLAcordLLCopper absorption and retention in young men at three levels of dietary copper using the stable isotope, 65CuAm J Clin Nutr19894958708782718922
- ScottKCTurnlundJRCompartment model of copper metabolism in adult menJ Nutr Biochem199457342350
- HornNTumerZMenkes disease and the occipital horn syndromeRoycePMSteinmannBConnective Tissue and its Heritable Disorders: Molecular, Genetic, and Medical AspectsNew York, NYJohn Wiley and Sons Inc2002651685
- LeeJPennaMMNoseYThieleDJBiochemical characterization of the human copper transporter Ctr1J Biol Chem200227764380438711734551
- FormigariAIratoPSantonAZinc, antioxidant systems and metallothionein in metal mediated-apoptosis: biochemical and cytochemical aspectsComp Biochem Physiol C Toxicol Pharmacol2007146444345917716951
- SpeiskyHGómezMBurgos-BravoFGeneration of superoxide radicals by copper-glutathione complexes: redox-consequences associated with their interaction with reduced glutathioneBioorg Med Chem20091751803181019230679
- HamzaIProhaskaJGitlinJDEssential role for Atox1 in the copper-mediated intracellular trafficking of the Menkes ATPaseProc Natl Acad Sci USA200310031215122012538877
- TurskiMLThieleDJNew roles for copper metabolism in cell proliferation, signaling, and diseaseJ Biol Chem2009284271772118757361
- CulottaVCYangMO’HalloranTVActivation of superoxide dismutases: putting the metal to the pedalBiochim Biophys Acta20061763774775816828895
- CamakarisJPetrisMJBaileyLGene amplification of the Menkes (MNK; ATP7A) P-type ATPase gene of CHO cells is associated with copper resistance and enhanced copper effluxHum Mol Genet1995411211721238589689
- MontyJFLlanosRMMercerJFKramerDRCopper exposure induces trafficking of the Menkes protein in intestinal epithelium of ATP7A transgenic miceJ Nutr2005135122762276616317117
- JankovRPBoerkoelCFHelimannJLethal neonatal Menkes’ disease with severe vasculopathy and fracturesActa Paediatr19988712129713009894833
- El MeskiniRCrabtreeKLClineLBMainsREEipperBARonnettGVATP7A (Menkes protein) functions in axonal targeting and synaptogenesisMol Cell Neurosci200734340942117215139
- SchliefMLWestTCraigAMHoltzmanDMGitlinJDRole of the Menkes copper- transporting ATPase in NMDA receptor-mediated neuronal toxicityProc Natl Acad Sci U S A200610340149191492417003121
- KalerSGGoldsteinDSHolmesCSalernoJAGahlWAPlasma and cerebrospinal fluid neurochemical pattern in Menkes diseaseAnn Neurol19933321711758434878
- KennersonMLNicholsonGAKalerSGMissense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathyAm J Hum Genet201086334335220170900
- QinZGongoraMCOzumiKRole of Menkes ATPase in angiotensin II-induced hypertension a key modulator for extracellular superoxide dismutase functionHypertension200852594595118768397
- RabikCAMaryonEBKaszaKShaferJTBartnikCMDolanMERole of copper transporters in resistance to platinating agentsCancer Chemother Pharmacol200964113314218998134
- WhiteCLeeJKambeTFritscheKPetrisMJA role for the ATP7A copper transporting ATPase in macrophage bactericidal activityJ Biol Chem200928449339493395619808669
- KimBETurskiMLNoseYCasadMRockmanHAThieleDJCardiac copper deficiency activates a systemic signaling mechanism that communicates with the copper acquisition and storage organsCell Metab201011535336320444417
- HornNCopper incorporation studies on cultured cells for prenatal diagnosis of Menkes’ diseaseLancet197630779701156115858201
- VergaVHallBKWangSRJohnsonSHigginsJVGloverTWLocalization of the translocation breakpoint in a female with Menkes syndrome to Xq13.2-q13.3 proximal to PGK-1Am J Hum Genet1991486113311382035533
- TumerZTommerupNTonnesenTKreuderJCraigIWHornNMapping of the Menkes locus to Xq13.3 distal to the X-inactivation center by an intrachromosomal insertion of the segment Xq13.3-q21.2Hum Genet19928866686721348049
- ChellyJTumerZTonnesenTIsolation of a candidate gene for Menkes disease that encodes a potential heavy metal binding proteinNat Genet19933114198490646
- MercerJALivingstonJHallBIsolation of a partial candidate gene for Menkes disease by positional cloningNat Genet19933120258490647
- VulpeCLevinsonBWhitneySPackmanSGitschierJIsolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPaseNat Genet1993317138490659
- StensonPDMortMBallEVThe human gene mutation database: 2008 updateGenome Med2009111319348700
- GuYHKodamaHMurataYATP7A gene mutations in 16 patients with Menkes disease and a patient with occipital horn syndromeAm J Med Genet200199321722211241493
- KalerSGLiewCJDonsanteAHicksJDSatoSGreenfieldJCMolecular correlates of epilepsy in early diagnosed and treated Menkes diseaseJ Inherit Metab Dis201033558358920652413
- TumerZMollerLBHornNScreening of 383 unrelated patients affected with Menkes disease and finding of 57 gross deletions in ATP7AHum Mutat200322645746414635105
- KalerSGInborn errors of copper metabolismHandb Clin Neurol20131131745175423622398
- TumerZAn overview and update of ATP7A mutations leading to Menkes disease and occipital horn syndromeHum Mutat201334341742923281160
- KalerSGMetabolic and molecular bases of Menkes disease and occipital horn syndromePediatr Dev Pathol199811859810463276
- DagenaisSLAdamANInnisJWGloverTWA novel frameshift mutation in exon 23 of ATP7A (MNK) results in occipital horn syndrome and not in Menkes diseaseAm J Hum Genet200169242042711431706
- MoizardMPRonceNBlessonSTwenty-five novel mutations including duplications in the ATP7A geneClin Genet201179324325321208200
- KalerSGATP7A-related copper transport diseases-emerging concepts and future trendsNat Rev Neurol201171152921221114
- KodomaHMurataYKobayashiMClinical manifestations and treatment of Menkes disease and its variantsPediatr Int199941442342910453199
- GrangeDKKalerSGAlbersGMPetterchakJAThorpeCMDeMelloDESevere bilateral panlobular emphysema and pulmonary arterial hypoplasia: unusual manifestations of Menkes diseaseAm J Med Genet A2005139215115516278898
- GuYHKodamaHKatoTCongenital abnormalities in Japanese patients with Menkes diseaseBrain Dev201234974674922361452
- MooreCMHowellRREctodermal manifestations in Menkes diseaseClin Genet19852865325404075564
- MizuguchiMItohMOzawaHMorikawaYA 2-year-old boy with hypoactivity of neonatal onset and profound developmental delayNeuropathology200727214514917494516
- DaishPWheelerEMRobertsPFJonesRDMenkes syndrome: report of a patient treated from 21 days of age with parenteral copperArch Dis Child19785312956958747401
- SasakiGIshiiTSatoSMultiple polypoid masses in the gastrointestinal tract in patient with Menkes disease on copper- histidinate therapyEur J Pediatr20041631274574615480778
- OkadaTSasakiFHondaSMiyagiHKubotaMTodoSMenkes disease with gastroesophageal reflux disease and successful surgical treatment: a case report and literatureTurk J Pediatr201052333333520718197
- ShiiharaTKatoMHonmaTProgressive sliding hiatal hernia as a complication of Menkes’ syndromeJ Child Neurol200217540140212150594
- SartorisDJLuzzattiLWeaverDDMacfarlaneJDHollisterDWParkerBRType IX Ehlers-Danlos syndrome. A new variant with pathognomonic radiographic featuresRadiology198415236656706463246
- KalerSGWestmanJABernesSMGastrointestinal hemorrhage associated with gastric polyps in Menkes diseaseJ Pediatr1993122193958419622
- JeongGUChoAHwangHA case of Menkes disease with unusual hepatomegalyKorean J Pediatr2008515538541
- RoycePMSteinmannBMarkedly reduced activity of lysyl oxidase in skin and aorta from a patient with Menkes disease showing unusually severe connective tissue manifestationsPediatr Res19902821371411975662
- RoycePMCamakarisJDanksDMReduced lysyl oxidase activity in skin fibroblast from patients with Menkes syndromeBiochem J198019225795866112984
- MenkesJHKinky hair disease: twenty five years laterBrain Dev198810277792839049
- MenkesJHKinky hair diseasePediatrics19725021811835045346
- GroverWDJohnsonWCHenkinRIClinical and biochemical aspects of trichopoliodystrophyAnn Neurol1979516571426469
- SmithVVAndersonGMaloneMSebireNJLight microscopic examination of scalp hair samples as an aid in the diagnosis of pediatric disorders: retrospective review of more than 300 cases from a single centreJ Clin Pathol200558121294129816311350
- WhitingDAStructural abnormalities of the hair shaftJ Am Acad Dermatol1987161 pt 11253805378
- KalerSGDiagnosis and therapy of Menkes syndrome, a genetic form of copper deficiencyAm J Clin Nutr199867suppl 5S1029S1034
- KalerSGHolmesCSGoldsteinDSDopamine beta-hydroxylase deficiency associated with mutations in a copper transporter geneAdv Pharmacol19984266689327848
- RobainOAubourgPRoutonMCDulacOPonsotGMenkes disease: a Golgi and electron microscopic study of the cerebellar cortexClin Neuropathol19887247523390973
- BinduPSTalyABKothariSElectro-clinical features and magnetic resonance imaging correlates in Menkes diseaseBrain Dev201335539840522921468
- Bahi-BuissonNKaminskaANabboutREpilepsy in Menkes disease: analysis of clinical stagesEpilepsia200647238038616499764
- JayawantSHalpinSWallaceSGMenkes kinky hair disease: an unusual caseEur J Pediatr Neurol200043131134
- FriedmanETHardenAKoivikkoMPampiglioneGMenkes’disease: neurophysiological aspectsJ Neurol Neurosurg Psychiatry197841650551097372
- WhiteSRReeseKSatoSKalerSGSpectrum of EEG findings in Menkes diseaseElectroencephalogr Clin Neurophysiol199387157617687955
- KalerSGHolmesCSGoldsteinDSNeonatal diagnosis and treatment of Menkes diseaseN Engl J Med2008358660561418256395
- BarnardROBestPVErdohaziMNeuropathology of Menkes’ diseaseDev Med Child Neurol1978205586597729906
- OkedaRGeiSChenIOkaniwaMShinomiyaMMatsubaraOMenkes’ kinky hair disease: morphological and immunohistochemical comparison of two autopsied patientsActa Neuropathol19918144504572028748
- ProudVKMussellHGKalerSGYoungDWPercyAKDistinctive Menkes disease variant with occipital horns: delineation of natural history and clinical phenotypeAm J Med Genet199665144518914740
- HermanTEMcAlisterWHBonifaceAWhyteMPOccipital horn syndromePediatr Radiol19922253633651408447
- DanksDMThe mild form of Menkes disease: progress report on the original caseAm J Med Genet19883038598643189408
- GerdesAMTønnesenTPergamentEVariability in clinical expression of Menkes syndromeEur J Pediatr198814221321353234433
- WestmanJARichardsonDCRennertOMMorrowG3rdAtypical Menkes steely hair diseaseAm J Med Genet19983038538583189407
- MøllerLBMogensenMHornNMolecular diagnosis of Menkes disease: genotype-phenotype correlationBiochimie200991101273127719501626
- TümerZLundCTolshaveJVuralBTønnesenTHornNIdentification of point mutations in 41 unrelated patients affected with Menkes diseaseAm J Hum Genet199760163718981948
- DasSLevinsonBWhitneySVulpeCPackmanSGitschierJDiverse mutations in patients with Menkes disease often lead to exon skippingAm J Hum Genet19945558838897977350
- KalerSGGahlWABerrySAHolmesCSGoldsteinDSPredictive value of plasma catecholamine levels in neonatal detection of Menkes diseaseJ Inherit Metab Dis19931659079088295415
- KalerSGTumerZPrenatal diagnosis of Menkes diseasePrenat Diagn19981832872899556046
- TønnesenTGerdesAMDamsgaardEFirst-trimester diagnosis of Menkes disease: intermediate copper values in chorionic villi from three affected male fetusesPrenat Diagn1989931591652710742
- VonkWIWijmengaCvan de SluisBRelevance of animal models for understanding mammalian copper homeostasisAm J Clin Nutr200888suppl 3S840S845
- LenartowiczMKrzeptowskiWLipinskiPMottled mice and non-mammalian models of Menkes diseaseFront Mol Neurosci201587226732058
- FraserASNayTGrowth of the mouse coat. II. Effect of sex and pregnancyAust J Biol Sci19536464565613126043
- HuntDMPrimary defect in copper transport underlies mottled mutants in the mouseNature19742494608528544858102
- TanakaKKobayashiKFujitaYFukuharaCOnosakaSMinKEffects of chelators on copper therapy of macular mouse, a model animal of Menkes’ kinky diseaseRes Commun Chem Pathol Pharmacol19906922172272396048
- KodamaHSatoEGuYHShigaKFujisawaCKozumaTEffect of copper and diethyldithiocarbamate combination therapy on the macular mouse, an animal model of Menkes diseaseJ Inherit Metab Dis200528697197816435190
- LenartowiczMKrzeptowskiWKotejaPChrzascikKMollerLBPrenatal treatment of mosaic mice (Atp7a mo-ms) mouse model for Menkes disease, with copper combined by dimethyldithiocarbamate (DMDTC)PLoS One201277e4040022815746
- MendelsohnBAYinCJohnsonSLWilmTPSolnica-KrezelLGitlinJDAtp7a determines a hierarchy of copper metabolism essential for notochord developmentCell Metab20064215516216890543
- MadsenECMorcosPAMendelsohnBAGitlinJDIn vivo correction of a Menkes disease model using antisense oligonucleotidesProc Natl Acad Sci U S A2008105103909391418316734
- NorgateMLeeESouthonAEssential roles in development and pigmentation for the Drosophila copper transporter DmATP7Mol Biol Cell200617147548416251357
- BahadoraniSBahadoraniPMarconEWalkerDWHillikerAJA Drosophila model of Menkes disease reveals a role for DmATP7 in copper absorption and neurodevelopmentDis Model Mech201031–2849120038716
- StaphanedeAGiorgiMAttilakosAPapadopoulosJTsiroudaMDinopoulosALate onset treatment on Menkes disease with a novel single nucleotide deletion in the ATP7A geneEur J Pediatr Neurol201519S121
- TchanMCWilchenBChristodoulouJThe mild form of Menkes disease: a 34 year progress report on the original caseJIMD Rep20139818423430551
- SanderCNiederhoffHHornNLife-span and Menkes kinky hair syndrome: report of a 13- year course of this diseaseClin Genet19883332282333359680
- KalerSGDasSLevinsonBSuccessful early copper therapy in Menkes disease associated with a mutant transcript containing a small in-frame deletionBiochem Mol Med199657137468812725
- ChristodoulouJDanksDMSarkarBEarly treatment of MD with parenteral copper -histidine: long-term follow-up of four treated patientsAm J Med Genet19987621541649511979
- KalerSGNeurodevelopment and brain growth in classic MD is influenced by age and symptomatology at initiation of copper treatmentJ Trace Elem Med Biol201428442743025281031
- PrasadANLevinSRuparCAPrasadCMenkes disease and infantile epilepsyBrain Dev2011331086687621924848
- TangJDonsanteADesaiVPatronasNKalerSGClinical outcomes in Menkes disease patients with a copper-responsive ATP7A mutation, G727RMol Genet Metab200895317418118752978
- SherwoodGSarkarBKortsakASCopper histidinate therapy in Menkes’ disease: prevention of progressive neurodegenerationJ Inherit Metab Dis19891223933962512453
- KalerSGMenkes disease mutations and response to early copper histidine treatmentNat Genet199613121228673098
- NassogneM-CSharradMHertz-PannierLMassive subdural haematomas in Menkes disease mimicking shaken baby syndromeChilds Nerv Syst2002181272973112483361
- JainPSharmaSSankhyanNMacrocephaly with diffuse white matter changes simulating a leukodystrophy in Menkes diseaseIndian J Pediatr201380216016222700386
- SahaSMridhaDAn unusual cause for focal convulsions: Menkes kinky hair diseaseJ Pediatr Neurol20131102123125