
Abstract
Purpose
Preventing opioid-induced hyperalgesia and tolerance continues to be a major clinical challenge, and the underlying mechanisms of hyperalgesia and tolerance remain elusive. Here, we investigated the role of sonic hedgehog (Shh) signaling in opioid-induced hyperalgesia and tolerance.
Methods
Shh signaling expression, behavioral changes, and neurochemical alterations induced by morphine were analyzed in male adult CD-1 mice with repeated administration of morphine. To investigate the contribution of Shh to morphine-induced hyperalgesia (MIH) and tolerance, Shh signaling inhibitor cyclopamine and Shh small interfering RNA (siRNA) were used. To explore the mechanisms of Shh signaling in MIH and tolerance, brain-derived neurotrophic factor (BDNF) inhibitor K252 and anti-BDNF antibody were used.
Results
Repeated administration of morphine produced obvious hyperalgesia and tolerance. The behavioral changes were correlated with the upregulation and activation of morphine treatment-induced Shh signaling. Pharmacologic and genetic inhibition of Shh signaling significantly delayed the generation of MIH and tolerance and associated neurochemical changes. Chronic morphine administration also induced upregulation of BDNF. Inhibiting BDNF effectively delayed the generation of MIH and tolerance. The upregulation of BDNF induced by morphine was significantly suppressed by inhibiting Shh signaling. In naïve mice, exogenous activation of Shh signaling caused a rapid increase of BDNF expression, as well as thermal hyperalgesia. Inhibiting BDNF significantly suppressed smoothened agonist-induced hyperalgesia.
Conclusion
These findings suggest that Shh signaling may be a critical mediator for MIH and tolerance by regulating BDNF expression. Inhibiting Shh signaling, especially during the early phase, may effectively delay or suppress MIH and tolerance.
Introduction
Opioid, such as morphine, plays an indispensable role in pain relief. However, long-term and repeated use of morphine no doubt leads to serious side effects, such as morphine-induced hyperalgesia (MIH) and tolerance.Citation1 MIH is characterized as a paradoxical increase of pain after long-term morphine use,Citation2 and tolerance is defined as a gradual loss of drug potency and reduced duration of action.Citation3 The two side effects usually emerged together. This phenomenon limits the beneficial therapeutic use of opioids in clinic. Thus, preventing and reversing MIH and tolerance is a clinical challenge. The mechanisms of MIH and tolerance are complex and involve many factors at different levels, such as the receptors, the cells, the ion channels, and the neural networks.Citation4–Citation7 However, despite decades of investigation, the specific cellular and molecular mechanisms underlying MIH and tolerance remain elusive.
Sonic hedgehog (Shh) is a secreted glycoprotein that plays a causal role in controlling the patterning of neural progenitor cells during development.Citation8 The components of Shh signaling in vertebrates mainly include Shh ligand, patched (Ptch) and smoothened (Smo) receptor, and Gli transcription factors.Citation8,Citation9 In the canonical pathway, activation of Shh signaling by binding of Shh to Ptc results in the activation of Smo and nuclear translocation of Gli.Citation10–Citation12 Emerging findings suggest that Shh plays important roles in the formation of neuronal circuits and synaptic plasticity.Citation8,Citation9 Shh could increase the size of presynaptic terminals and the frequency of miniature excitatory postsynaptic currents at hippocampal neuron synapses.Citation13,Citation14 Although the role of Shh during neurodevelopment is well addressed, the effect of Shh in adults remains unclear. It was reported that activation of Shh signaling was induced by acute brain injuryCitation15 and mediated brain plasticity.Citation16 Recently, it was reported that Shh signaling was involved in nociceptive regulation.Citation17–Citation19 Shh mutation resulted in lack of nociceptive sensitization in drosophila.Citation18 Besides, inhibited Shh signaling significantly suppressed complete Freund’s adjuvant-induced thermal analgesia and sciatic nerve ligation-induced mechanical allodynia.Citation18 More recently, it was found that Shh signaling contributed to pancreatic cancer pain.Citation19 Accumulating evidence suggests an involvement of a similar molecular mechanism underlying the nociceptive pain in MIH and tolerance,Citation20,Citation21 but whether Shh signaling is involved in MIH and tolerance remains unclear.
In the present study, we provide integrated evidence that activation of Shh signaling contributed to the generation of MIH and tolerance by upregulating brain-derived neurotrophic factor (BDNF) expression. Inhibiting Shh signaling could effectively prevent MIH and tolerance.
Materials and methods
Animals
Adult male CD-1 mice (24–28 g) were purchased from Shanghai Experimental Animal Center of Chinese Academy of Science. The mice were housed in a temperature-controlled environment with 12-hour light–dark cycles and were fed standard laboratory diet and water ad libitum. All mice were handled daily for 5 days before the start of the experiment to minimize the stress reaction to manipulation. The experimental protocols were approved by the Animal Research Committee of Xuzhou Medical University and were in accordance with the Declaration of the National Institutes of Health Guide for Care and Use of Laboratory Animals (Publication No. 85-23.)
Drugs and anesthesia
Mice were anesthetized with pentobarbital (50 mg/kg, intraperitoneally [i.p.]) for preparing the spinal cord tissue for Western blotting and immunobiochemistry. Shh signaling inhibitor cyclopamine and activator smoothened agonist (SAG) were purchased from Selleck (Houston, TX, USA). Tyrosine kinase inhibitor K252, BDNF inhibitor, was purchased from Biomol (Plymouth Meeting, PA, USA). Each of these drugs was dissolved in PBS ordimethyl sulfoxide (DMSO) and diluted in PBS (final concentration of DMSO was 1%). Cyclopamine (10 mg/kg), SAG (5 mg/kg), and the vehicle controls were injected i.p. (1 mL each). K252 (80 μg/kg) and anti-BDNF antibody (2 μg/kg) were injected intrathecally (i.t.). To confirm the role of Shh in MIH, Shh siRNA (Santa Cruz Biotechnology Inc., Dallas, TX, USA) was injected (1 μg/10 μL, i.t.) by means of lumbar puncture at the intervertebral space of L4–L5.
MIH model and behavior test
To construct the animal model of MIH, mice were administered morphine repeatedly (10 mg/kg, i.p.) twice a day for 7 consecutive days. Mice in the sham group were administered the same dose of saline (1 mL, i.p.) at the same time points. The anti-nociceptive effect was measured 30 minutes after each injection of morphine. Morphine tolerance was tested by hot plate. Mice were placed on a 55°C hot plate apparatus, and the latency to lick a paw was measured. In order to avoid empyrosis, the maximal latency was set as 60 seconds. Data were calculated as percentage maximal possible effect (MPE%), which was calculated by the following formula: MPE% = [(drug response time – basal response time)]/(30 seconds – basal response time)]×100. Thermal hyperalgesia was assessed by an analgesia meter (IITC Model 336 Analgesia Meter, Series 8; IITC Life Science Inc.; Woodland Hills, CA, USA). In brief, animals were placed in a box with a temperature-controlled glass floor. The heat source was focused on a portion of the hindpaw, which was flush against the glass, and a radiant thermal stimulus was delivered to that site. The stimulus shut off when the hindpaw moved (or after 20 seconds to prevent tissue damage). Thermal stimuli were delivered three times to each hindpaw at 5–6 minute intervals. To explore the role of Shh in MIH generation, cyclopamine was injected 15 minutes before morphine treatment twice a day from day 1 to day 3. To investigate the effect of Shh on MIH maintenance, cyclopamine was injected for 3 consecutive days after the MIH model was established, from day 5 to day 7, twice a day.
Western blotting
To identify temporal expression of Shh signaling (Shh, Ptch1, Smo, Gli1), whole-cell protein extract lysates were used. To identify the activation of Shh signaling, nuclear extracts were prepared using an NE-PER Nuclear and Cytoplasmic Extraction Kit (Pierce Biotechnology, Rockford, IL, USA) according to the manufacturer’s instruction. L1–L6 spinal cord segments and L4–L6 dorsal root ganglions (DRGs) were quickly removed from deeply anesthetized mice and homogenized in ice-cold radio-immunoprecipitation assay lysis buffer containing a cocktail of protease inhibitors. Total proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to 0.2 μm polyvinylidenefluoride membrane. The following primary antibodies were used: anti-Shh (1:200; Santa Cruz Biotechnology Inc.), anti-Ptch1 (1:1000; Sigma , St. Louis, MO, USA), anti-Smo (1:1000; Abcam , Cambridge, UK), anti-Gli1 (1:200; Santa Cruz Biotechnology Inc.), anti-BDNF (1:1000; Abcam), anti-Histone H3 (1:1000; Abcam), anti-c-fos (1:500; Cell Signaling Tech, Danvers, MA, USA), anti-CGRP (1:1000; Abcam), and anti-GAPDH (1:10000; Sigma). The filters were developed using ECL reagents (PerkinElmer Inc., Waltham, MA, USA) with secondary antibodies from Millipore Bioscience Research Reagents (EMD Millipore, Billerica, MA, USA). Data were analyzed with a Molecular Imager (Gel DocTM XR, 170-8170; Bio-Rad Laboratories Inc., Hercules, CA, USA) and the associated software Quantity One-4.6.5 (Bio-Rad Laboratories Inc.).
Immunohistochemistry
Under deep anesthesia, mice were transcardially perfused with PBS followed by 4% paraformaldehyde with 1.5% picric acid in 0.16 M PBS (pH 7.2–7.4), and then the L4–L6 lumbar segment and L4–L6 DRGs were dissected out and postfixed in the same fixative overnight. The embedded blocks were sectioned as 30 μm thick and processed for immunofluorescence. Sections from each group (five mice in each group) were incubated with the following primary antibodies: anti-Shh (1:100, Santa Cruz Biotechnology Inc.), anti-c-Fos (1:100, Santa Cruz Biotechnology Inc.), and anti-CGRP (1:1000, EMD Millipore), respectively. Then the sections were washed in 50 mM Tris-HCl (pH 7.4) PBS for 5 minutes three times and incubated in the secondary antibody for 2 hours at room temperature. After washing three times in PBS, the sections were observed by a confocal microscope (Leica TCS SPEII; Leica Microsystems, Wetzlar, Germany) for morphologic details. Images were analyzed using MicroSuite image analysis software. For double staining, the same procedure was performed to the second primary and secondary antibodies. Sections were mounted on slides and covered with 90% glycerin for observation under a confocal microscope. The dilution of antibodies used included anti-Shh (1:100; Santa Cruz Biotechnology Inc.), anti-Gli1 (1:100; Santa Cruz Biotechnology Inc.), and anti-BDNF (1:200; Abcam). To obtain the quantitative measurements of CGRP immunofluorescence, 15–20 fields covering the entire dorsal horn in each group were evaluated and photographed at the same exposure time to generate the raw data. The average green fluorescence intensity of each pixel was normalized to the background intensity in the same image. Tissues were collected at days 4 and 7 after the first injection of morphine.
Enzyme-linked immunosorbent assay (ELISA)
Under deep anesthesia, the L1–L6 spinal cord segments and L4–L6 DRGs of mice were rapidly removed and homogenized in ice-cold 0.01 mol/L PBS. Protein concentrations were determined by BCA protein assay. The level of Shh concentration was measured by ELISA using Sonic Hedgehog N-Terminus Quantikine ELISA Kit (R&D Systems, Inc., Minneapolis, MN, USA), according to the manufacturer’s instruction.
Statistical analysis
SPSS 16 (SPSS Inc., Chicago, IL, USA) was used to conduct all the statistical analyses. Alteration of expression of the proteins detected and the behavioral responses to MIH were analyzed with one-way analysis of variance (ANOVA), and the differences in withdrawal threshold over time among groups were tested with two-way ANOVA with repeated measures, followed by Bonferroni post hoc test. All data are presented as mean ± standard error of the mean (SEM). Statistical results were considered significant if P<0.05.
Results
Inhibiting Shh signaling delayed and suppressed tolerance and MIH generation, but showed no effect on the maintenance of tolerance and MIH
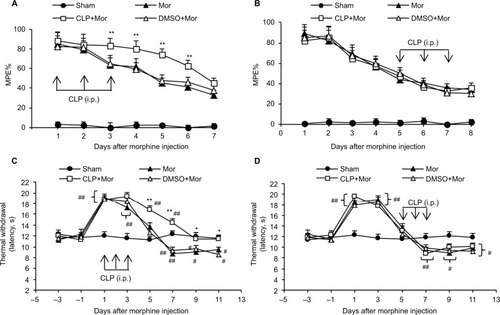
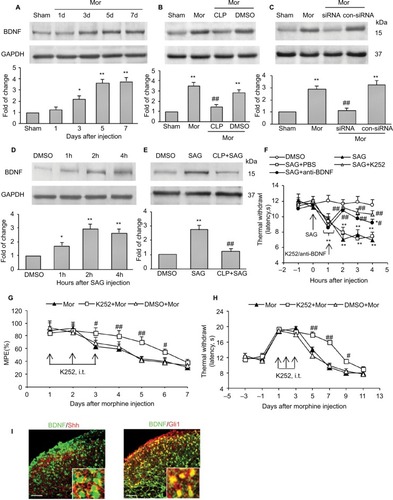
To address whether Shh signaling was involved in the regulation of morphine tolerance, we first conducted behavioral tests by using Shh signaling inhibitor cyclopamine. As shown in , mice that received chronic morphine treatment showed significant decrease of MPE% (), which refers to morphine tolerance, from day 3 after morphine administration. Besides, with morphine treatment, mice showed obvious thermal hyperalgesia from day 5 after morphine injection, lasting at least to day 11 (). Repetitive treatment with cyclopamine 15 minutes before morphine injection (10 mg/kg, i.p., twice a day) at days 1, 2, and 3 significantly delayed and suppressed morphine-induced tolerance () and thermal hyperalgesia () (P<0.05). On the contrary, the same dose of cyclopamine injected at days 5, 6, and 7 failed to reverse or suppress morphine-induced tolerance () and thermal hyperalgesia () (P>0.05). The normal pain sensation was not altered in the sham group. Taken together, these results demonstrated that Shh signaling may be involved in the regulation of tolerance and MIH generation, but not the maintenance of tolerance and MIH.
Figure 1 Effects of cyclopamine on morphine-induced hyperalgesia and tolerance.
Abbreviations: i.p., intraperitoneally; MPE%, percentage of maximal possible effect; Mor, morphine; CLP, cyclopamine; DMSO, dimethyl sulfoxide.
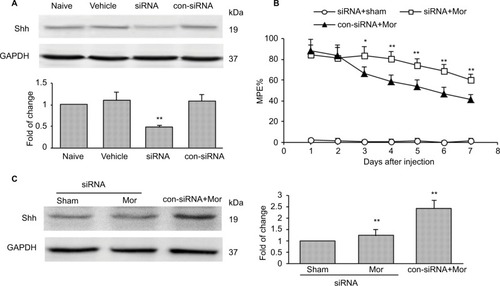
To confirm the role of Shh signaling in MIH and tolerance, Shh siRNA was used. Shh siRNA (1 μg/10 μL) was injected i.t. 3 consecutive days before morphine administration. Western blotting showed that spinal administration of Shh siRNA for 3 consecutive days effectively decreased the expression of Shh in spinal cord (). Furthermore, behavioral tests showed that with Shh gene knockdown, the reduction of morphine-induced MPE% () was significantly delayed and suppressed. The normal pain sensation showed no obvious changes in the siRNA + sham group, and control siRNA (con-siRNA) showed no effect on tolerance (). In addition, siRNA administration significantly inhibited morphine treatment-induced Shh protein upregulation in spinal cord, while con-siRNA showed no effect on Shh expression ().
Figure 2 Knockdown of Shh in spinal cord prevents the reduction of chronic morphine treatment-induced MPE%.
Abbreviations: MPE%, percentage of maximal possible effect; siRNA, small interfering RNA; i.t., intrathecally; Mor, morphine; con-siRNA, control-siRNA.
Inhibiting Shh signaling suppressed chronic morphine treatment-induced neurochemical signs in spinal cord
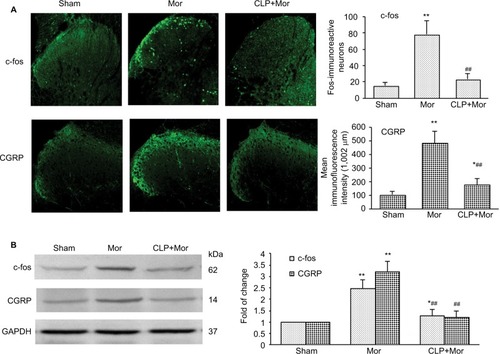
It was reported that chronic morphine exposure could cause neurochemical alterations in spinal cord, including c-fos and CGRP.Citation22–Citation25 Consistent with previous studies, both immunofluorescence staining () and Western blot test () showed that with repetitive administration of morphine, the expressions of c-fos and CGRP significantly increased in the spinal cord of the mice. Pretreatment with cyclopamine to inhibit Shh signaling significantly delayed or suppressed the upregulation of c-fos and CGRP ().
Figure 3 Shh signaling inhibition with cyclopamine suppressed morphine treatment-induced upregulation of c-fos and CGRP in the spinal cord of mice.
Abbreviations: i.p., intraperitoneally; Mor, morphine; CLP, cyclopamine.
Shh signaling upregulated and activated following repeated morphine treatment
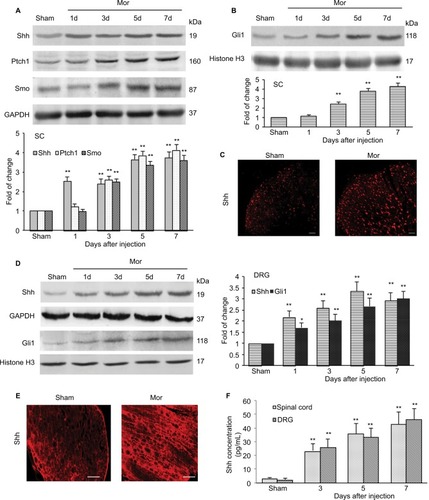
Given that inhibiting Shh signaling can delay or suppress morphine exposure-induced behavioral and neurochemical changes, we examined the expression and activity of Shh signaling in spinal cord under the same conditions. Western blotting showed that repetitive administration of morphine induced a time-dependent and long-lasting increase in the expressions of Shh and its receptor Ptch1 and Smo in spinal cord, which began on postinjection day 3. The upregulation of Shh signaling remained until day 7 (the last examination) (). Gli1 is a nuclear transcription factor and will transfer to the nucleus after the activation of Shh signaling.Citation8,Citation9 In order to prove the activation of Shh signaling, the expression of Gli1 in nuclear extract was tested. It was shown that the expression of Gli1 in the nucleus was also increased significantly after repetitive administration of morphine, from day 3 after morphine injection (). These results suggested that repeated morphine treatment could increase and activate Shh signaling in spinal cord. The immunofluorescence staining also showed that after morphine treatment, the expression of Shh was significantly increased in the dorsal horn of the spinal cord (). Furthermore, we also investigated the expression and activation of Shh signaling in DRG. Both the Western blot analysis () and immunofluorescence staining () showed that, after chronic morphine treatment, the expression of Shh protein and the nuclear translocation of Gli1 were significantly increased in a time-dependent pattern. Besides, immunofluorescence staining indicated that the increased Shh proteins were primarily located in small and medium neurons (). ELISA showed that the concentration and release of Shh protein was significantly increased in DRG and spinal cord after morphine treatment (). Taken together, these results suggested that Shh signaling was significantly upregulated and activated in DRG and spinal cord during morphine tolerance.
Figure 4 Expression and activation of Shh signaling after chronic morphine treatment.
Abbreviations: SC, spinal cord; DRG, dorsal root ganglion; i.t., intrathecally; Mor, morphine; ELISA, enzyme linked immunosorbent assay.
Activation of Shh signaling regulating the expression of BDNF in spinal cord during MIH and tolerance
To explore the mechanisms by which Shh signaling induces MIH and tolerance, we first examined the expression of BDNF, which is reported as the important mediator for MIH and tolerance.Citation26,Citation27 Consistent with previous studies, we found that the expression of BDNF increased significantly after chronic morphine treatment. It increased from day 3 after morphine administration and lasted at least 5 days until final detection (). Repetitive administration of cyclopamine (10 mg/kg, i.p., at days 1, 2, and 3) significantly inhibited morphine-induced upregulation of BDNF (). Similarly, genetic knockdown of Shh with Shh siRNA also significantly inhibited increase in morphine-induced BDNF (). In naïve mice, intraperitoneal injection of SAG (5 mg/kg), Shh signaling activator, caused a rapidly increased (within 1 hours) expression of BDNF protein (), and this effect of SAG on BDNF expression was prevented by cyclopamine pre-administration (). SAG injection can also induce thermal hyperalgesia in naïve mice (). Pretreatment with K252 (80 μg/kg, i.t.), BDNF inhibitor, or anti-BDNF antibody (2 μg/kg, i.t.), significantly prevented and suppressed SAG-induced hyperalgesia (). Besides, repetitive injection of K252 (80 μg/kg, i.t.) also effectively prevented and delayed the generation of tolerance () and MIH (). Furthermore, to confirm BDNF was under regulated by Shh signaling during morphine tolerance, immunofluorescence double staining was used to investigate the location of BDNF. It was shown that, in the dorsal horn of spinal cord, increased BDNF was colocalized with Shh (, left) and Gli1 (, right). These results confirm that it was indeed possible that BDNF was regulated by Shh signaling. Taken together, these results indicated that the upregulation of BDNF in the spinal cord after chronic morphine administration may be induced by Shh signaling activation and that Shh signaling contributes to MIH and tolerance by regulating BDNF expression.
Figure 5 Shh signaling contributed to tolerance and MIH through regulating BDNF expression.
Abbreviations: MIH, morphine-induced hyperalgesia; BDNF, brain-derived neurotrophic factor; siRNA, small interfering RNA; i.p., intraperitoneally; i.t., intrathecally; Mor, morphine; CLP, cyclopamine; DMSO, dimethyl sulfoxide; SAG, smoothened agonist; MPE, maximal possible effect.
Discussion
In our present study, Shh was identified as an important molecular signal for the regulation of MIH and tolerance. Activation of Shh signaling is involved in the development of MIH and tolerance, and inhibition or downregulation of Shh signaling significantly delayed and alleviated MIH and tolerance. The possible mechanism of Shh on MIH may be mediated by BDNF. To our knowledge, this is the first demonstration of a regulatory role of Shh signaling in MIH and tolerance through BDNF.
MIH and tolerance to morphine is one of the major side effects associated with its long-term administration. This phenomenon limits the beneficial therapeutic use of opioids. Many attempts have been made to find agents that can prevent this adverse effect. Although MIH and tolerance are not fully understood, many potential mechanisms have been identified for this process, including sensitization of the nerve system and synaptic plasticity.Citation21 The withdrawal from opioids, such as morphine, fentanyl, and remifentanyl, induced a potentiation of the synapse between nociceptive C fibers and neurons in the superficial dorsal horn of the spinal cord.Citation28,Citation29
Shh is a secreted protein that controls the patterning of neural progenitor cells, and their neuronal and glial progeny, during development. Emerging findings suggest that Shh also has important roles in the formation and plasticity of neuronal circuits in hippocampus.Citation8 It was reported that Shh is present in both pre- and postsynaptic terminals, where it may be associated with synaptic vesicles and endosomes.Citation14 Besides, activation of Shh signaling can also increase the size of presynaptic terminals at hippocampal neuron synapses.Citation13 The ability of the Shh signaling pathway to regulate neuronal connectivity and synaptic plasticity both during and after the development has been observed in many parts of the nervous system.Citation30–Citation32 Recently, Shh signaling was reported to be required for modulating nociception and analgesia.Citation18,Citation19,Citation33 The expression of Shh significantly increased after sciatic nerve injury and lasted at least 4 weeks,Citation17 which is consistent with pain-related behavioral changes. Inhibiting Shh signaling by cyclopamine effectively attenuated neuropathic painCitation18 and decreased the concentration of cytokines, such as tumor necrosis factor-α and interleukin-1β.Citation34 In our present study, we also found that, after repetitive administration of morphine, the expression of Shh and its related proteins significantly increased in both spinal cord and DRG in a time-dependent manner. And, the expression and activation of Shh signaling were consistent with MIH and tolerance in the time course. The increased Shh was located not only in laminae I and II of the dorsal horn but also in deeper dorsal horn (laminae VI-V), which receive Aδ nociceptor project. There are two major classes of nociceptive afferents; Aδ fibers and C fibers. Aδ nociceptor projects to lamina I as well as to deeper dorsal horn (lamina VI-V). C nociceptors project more superficially to laminae I and II. Pharmacological and genetic blocking of Shh signaling successfully rescued the analgesic effect of morphine and delayed the generation of MIH and tolerance. Meanwhile, the increased levels of both c-fos and CGRP following repetitive administration of morphine were also inhibited by blocking Shh signaling. These data strongly suggested that Shh signaling is involved in MIH and tolerance. Here we chose to focus on chronic morphine tolerance, rather than acute morphine tolerance, because it resembles more closely clinical practice.
Although the present data indicated that Shh may be involved in the regulation of MIH and tolerance, it will also be important to determine how Shh signaling regulates them. Previous studies performed in the cavernous nerveCitation35 and in the sciatic nerveCitation17 suggest that BDNF could be a target of Shh.Citation36 Activation of Shh pathway upregulates BDNF to protect the cortical neurons against oxidative stress.Citation37 BDNF is a neurotrophic factor. It is implicated in neuronal plasticity and synaptic function.Citation38,Citation39 Recently, BDNF was found to be involved in MIH. Upregulation of BDNF expression by DNA methylation inhibitor augmented MIH.Citation40 In our present study, we found that after repeated morphine administration, the expression of BDNF in spinal cord increased significantly. Pharmacological and genetic blocking of Shh signaling effectively suppressed the increase of BDNF induced by MIH. Furthermore, we also found that in naïve mice, after SAG (Shh signaling activator) administration, the expression of BDNF increased obviously. Besides, the hyperalgesia induced by SAG injection could be significantly suppressed and reversed by BDNF inhibitor and anti-BDNF antibody. Also, immunofluorescence double staining showed that increased BDNF was colocalized with Shh and Gli1 in the dorsal horn of spinal cord. Taken together, these data suggested that BDNF may be downstream of Shh signaling during MIH. Nevertheless, the possible mechanism of how the Shh signaling induced the upregulation of BDNF needs to be further studied. After chronic morphine treatment, Shh signaling was upregulated and activated in DRG and spinal cord. Activated Shh signaling induced nociceptive neuron hyperexcitability and nociceptive inputs increased by upregulating BDNF expression, and then induced morphine tolerance and MIH.
Interestingly, there was an unexpected finding that Shh signaling inhibitor showed no effect on the maintenance of MIH and tolerance. Administration of cyclopamine at days 5, 6, and 7 after morphine injection, when MIH and tolerance had already emerged, showed no effect on MPE% and thermal withdrawal latency decrease. This is an interesting finding. This result demonstrated that the mechanisms of MIH and tolerance generation may be different from those of maintenance, and Shh signaling activation may be the trigger of MIH and tolerance generation.
Conclusion
The present study suggested a new clinical strategy for treating and preventing MIH and tolerance. Shh signaling in spinal cord may play a key role in chronic morphine tolerance and MIH by regulating BDNF expression. Inhibiting Shh signaling, especially during the early phase of MIH and tolerance, may effectively delay or suppress MIH and tolerance.
Author contributions
SL, J-LY, and X-XW designed the studies. J-LY performed the molecular biological test. X-XW and SM performed the animal behavioral tests. Z-JS, YZ, and X-LW performed data collection and statistical analysis. SL and Y-PL oversaw the execution of the project. SL, J-LY, and X-XW interpreted the results and drafting the manuscript. All the authors discussed the results and revised the manuscript critically for important intellectual content.
Acknowledgments
The authors thank Xue-jun Song, MD, PhD, and Zhi-jiang Huang, PhD, (Department of Neurobiology, Parker University, Dallas, TX, 75229, USA) for their help in this study. This work was supported by a grant from the National Natural Science Foundation of China (NSFC-81371242, NSFC-81671084), Qing Lan Project of Jiangsu province, Nature Science Foundation of Jiangsu province (BK20161175), and “Six One” Project of Jiangsu province (LGY2016039).
Disclosure
The authors report no conflicts of interest in this work.
References
- McquayHJOpioids in pain managementLancet19993532229223210393001
- HuaZLiuLShenJMesenchymal stem cells reversed morphine tolerance and opioid-induced hyperalgesiaSci Rep201663209627554341
- WangJXuWZhongTmiR-365 targets β-arrestin 2 to reverse morphine tolerance in ratsSci Rep201663828527922111
- HayashiYMorinagaSZhangJBK channels in microglia are required for morphine-induced hyperalgesiaNat Commun201671169727241733
- CheppudiraBPTrevinoAVPetzLNAnti-nerve growth factor antibody attenuates chronic morphine treatment-induced tolerance in the ratBMC Anesthesiol2016167327596139
- SanchezblazquezPRodriguezmunozMBerrocosoEThe plasticity of the association between mu-opioid receptor and glutamate ionotropic receptor N in opioid analgesic tolerance and neuropathic painEur J Pharmacol20137169410523499699
- MartiniLWhistlerJLThe role of mu opioid receptor desensitization and endocytosis in morphine tolerance and dependenceCurr Opin Neurobiol20071755656418068348
- YaoPJPetraliaRSMattsonMPSonic hedgehog signaling and hip-pocampal neuroplasticityTrends Neurosci20163984085027865563
- LucaADCerratoVFucàESonic hedgehog patterning during cerebellar developmentCell Mol Life Sci20167329130326499980
- InghamPWMcmahonAPHedgehog signaling in animal development: paradigms and principlesGenes Dev2001153059308711731473
- RuelLRodriguezRGalletAStability and association of Smoothened, Costal2 and Fused with Cubitus interruptus are regulated by hedgehogNat Cell Biol2003590791314523402
- BriscoeJSmallSMorphogen rules: design principles of gradient-mediated embryo patterningDevelopment20151423996400926628090
- MitchellNPetraliaRSCurrierDGSonic hedgehog regulates presynaptic terminal size, ultrastructure and function in hippocampal neuronsJ Cell Sci20121254207421322641692
- PetraliaRSWangYMattsonMPYaoPJSonic hedgehog distribution within mature hippocampal neuronsCommun Integr Biol2011477577722446553
- AmankulorNHambardzumyanDPyonteckSMBecherOJJoyceJAHollandECSonic hedgehog pathway activation is induced by acute brain injury and regulated by injury-related inflammationJ Neurosci200929102991030819692604
- DingXLiYLiuZThe sonic hedgehog pathway mediates brain plasticity and subsequent functional recovery after bone marrow stromal cell treatment of stroke in miceJ Cereb Blood Flow Metab2013331015102423549381
- HashimotoMIshiiKNakamuraYNeuroprotective effect of sonic hedgehog up-regulated in Schwann cells following sciatic nerve injuryJ Neurochem200810791892718786173
- BabcockDTShiSJoJShawMGutsteinHBGalkoMJHedgehog signaling regulates nociceptive sensitizationCurr Biol2011211525153321906949
- HanLMaJDuanWPancreatic stellate cells contribute pancreatic cancer pain via activation of sHH signaling pathwayOncotarget20167181461815826934446
- MayerDJMaoJHoltJPriceDDCellular mechanisms of neuropathic pain, morphine tolerance, and their interactionsProc Natl Acad Sci U S A1999967731773610393889
- RoeckelLALe CozGMGavériaux-RuffCSimoninFOpioid-induced hyperalgesia: cellular and molecular mechanismsNeuroscience201633816018227346146
- YanHYuLExpression of calcitonin gene-related peptide receptor subunits in cultured neurons following morphine treatmentNeurosci Lett2013544525523570731
- WangZChabotJQuirionROn the possible role of ERK, p38 and CaMKII in the regulation of CGRP expression in morphine-tolerant ratsMol Pain201176821933441
- HaoSMataMGoinsWFGloriosoJCFinkDJTransgene-mediated enkephalin release enhances the effect of morphine and evades tolerance to produce a sustained antiallodynic effect in neuropathic painPain200310213514212620604
- LiuSLiuWLiuYBlocking EphB1 receptor forward signaling in spinal cord relieves bone cancer pain and rescues analgesic effect of morphine treatment in rodentsCancer Res2011714392440221555368
- WenYRTanPHChengJKLiuYCJiRRMicroglia: a promising target for treating neuropathic and postoperative pain, and morphine toleranceJ Formos Med Assoc201111048749421783017
- HuXMCaoSBZhangHLDownregulation of miR-219 enhances brain-derived neurotrophic factor production in mouse dorsal root ganglia to mediate morphine analgesic tolerance by upregulating CaMKIIγMol Pain2016121467014683
- DrdlaRGassnerMGinglESandkühlerJInduction of synaptic long-term potentiation after opioid withdrawalScience200932520721019590003
- Drdla-SchuttingRBenrathJWunderbaldingerGSandkühlerJErasure of a spinal memory trace of pain by a brief, high-dose opioid administrationScience201233523523822246779
- ChouYZhengXBeachyPALuoLPatterning axon targeting of olfactory receptor neurons by coupled hedgehog signaling at two distinct stepsCell201014295496620850015
- YamPTCharronFSignaling mechanisms of non-conventional axon guidance cues: the Shh, BMP and Wnt morphogensCurr Opin Neurobiol20132396597324183376
- YaoPJPetraliaRSOttCWangYXLippincott-SchwartzJMattsonMPDendrosomatic sonic hedgehog signaling in hippocampal neurons regulates axon elongationJ Neurosci201535161261614126658865
- MoreauNMauborgneABourgoinSEarly alterations of Hedgehog signaling pathway in vascular endothelial cells after peripheral nerve injury elicit blood-nerve barrier disruption, nerve inflammation, and neuropathic pain developmentPain201615782783926655733
- LiRCaiLDingJHuCMWuTNHuXYInhibition of hedgehog signal pathway by cyclopamine attenuates inflammation and articular cartilage damage in rats with adjuvant-induced arthritisJ Pharm Pharmacol20156796397125645065
- BondCWAngeloniNHarringtonDAStuppSPodlasekCASonic hedgehog regulates brain-derived neurotrophic factor in normal and regenerating cavernous nervesJ Sex Med20131073073723237228
- HeWCuiLZhangCZhangXHeJXieYSonic hedgehog promotes neurite outgrowth of primary cortical neurons through up-regulating BDNF expressionNeurochem Res20154168769526459360
- DaiRZhuSXiaYSonic hedgehog protects cortical neurons against oxidative stressNeurochem Res201036677520848190
- AntalAChaiebLMoliadzeVBrain-derived neurotrophic factor (BDNF) gene polymorphisms shape cortical plasticity in humansBrain Stimul2010323023720965453
- CastilloDVEscobarMLA role for MAPK and PI-3K signaling pathways in brain-derived neurotrophic factor modification of conditioned taste aversion retentionBehav Brain Res201121724825220974194
- ChaoYXieFLiXDemethylation regulation of BDNF gene expression in dorsal root ganglion neurons is implicated in opioid-induced pain hypersensitivity in ratsNeurochem Int201697919826970395