Abstract
Two decades of investigations have failed to unequivocally clarify the functions and the molecular nature of imidazoline-2 receptors (I2R). However, there is robust pharmacological evidence for the functional modulation of monoamino oxidase (MAO) and other important enzyme activities by I2 site ligands. Some compounds of this class proved to be active experimental tools in preventing both experimental pain and opioid tolerance and dependence. Unfortunately, even though these compounds bind with high potency to central I2 sites, they fail to represent a valid clinical opportunity due to their pharmacokinetic, selectivity or side-effects profile. This paper presents the preclinical profile of a novel I2 ligand (2-phenyl-6-(1H-imidazol-1yl) quinazoline; [CR4056]) that selectively inhibits the activity of human recombinant MAO-A in a concentration-dependent manner. A sub-chronic four day oral treatment of CR4056 increased norepinephrine (NE) tissue levels both in the rat cerebral cortex (63.1% ±4.2%; P < 0.05) and lumbar spinal cord (51.3% ± 6.7%; P < 0.05). In the complete Freund’s adjuvant (CFA) rat model of inflammatory pain, CR4056 was found to be orally active (ED50 = 5.8 mg/kg, by mouth [p.o.]). In the acute capsaicin model, CR4056 completely blocked mechanical hyperalgesia in the injured hind paw (ED50 = 4.1 mg/kg, p.o.; ED100 = 17.9 mg/kg, p.o.). This effect was dose-dependently antagonized by the non-selective imidazoline I2/α2 antagonist idazoxan. In rat models of neuropathic pain, oral administration of CR4056 significantly attenuated mechanical hyperalgesia and allodynia. In summary, the present study suggests a novel pharmacological opportunity for inflammatory and/or neuropathic pain treatment based on selective interaction with central imidazoline-2 receptors.
Introduction
Imidazoline binding sites, also known as imidazoline receptors (IRs), are widely distributed in mammalian cells of the central (CNS) and peripheral (PNS) nervous systems, liver, kidney and heart.Citation1 To date, three subtypes of such proteins have been proposed (I1, I2, I3). However, only in the case of the I1 subtype was a complete molecular characterization achieved. This protein is also referred to as IRAS (imidazoline receptor antisera-selected protein)Citation2 and is involved in clonidine-like hypotensive activity.Citation3
Conversely, imidazoline-2 binding sites (I2Rs) are still orphan proteins from a molecular point of view, although there is a great deal of experimental evidence related to functional modulations by I2R ligands of monoamino-oxidase (MAO) activity through their interaction with accessory sites present on discrete populations of MAO enzymes.Citation4–Citation7 Other purported activities of I2R ligands are related to the modulation of brain creatine kinase (B-CK)Citation8 and semicarbazide-sensitive amino-oxidase (SSAO).Citation9 The physiological relevance of these molecular targets may suffice to explain the putative involvement of I2R in various diseasesCitation10–Citation12 including neuropathic and inflammatory pain.Citation13–Citation16
A number of experimental studies have evaluated the interaction between I2R ligands and the opioid system. Agmatine, the putative endogenous agonist of imidazoline receptors,Citation17 and 2-(2-benzofuranyl)-2-imidazoline (2-BFI), a potent and selective I2 ligand,Citation18 have been shown to modulate the analgesic effect of morphine in rodents and block the development of tolerance to morphine. Ruiz-Durantez et alCitation18 reported an important cross-talk between I2R ligands and the opioid system in the locus coeruleus. Since the descending noradrenergic pathway originates in the locus coeruleus and extends to the dorsal horn of the spinal cord, such interaction could have significant relevance in controlling the central sensitization from higher centres to spinal nociceptors. These results suggest that compounds of this class, being able to cross the blood–brain barrier efficiently, may directly control the descending pain pathway, and attenuate the development of opioid tolerance and dependence. This could be useful in the treatment of chronic and neuropathic pain.
Neuropathic pain is a chronic pain condition often refractory to multiple treatment modalities including opioids and non-steroidal anti-inflammatory drugs (NSAIDs). Thus, current treatment options in neuropathic pain conditions are limited to some tricyclic antidepressants (TCAs), anticonvulsants and systemic local anesthetics. Unfortunately, clinical data demonstrate that these drugs usually provide either a limited efficacy or low levels of compliance due to their side effect profile.Citation19,Citation20
In the present study, we characterized the preclinical pharmacological effects of a novel I2R ligand (laboratory code CR4056), and tested the hypothesis that this compound induces anti-nociceptive effects in rat models of experimental pain.
Material and methods
Animals
Male Wistar rats (Harlan, Bresso, Italy) weighing 200–300 g were used for all experiments. Animals were housed in plastic cages, with free access to standard laboratory food and water, under controlled conditions of lighting, humidity, and temperature. All experiments were in compliance with the European Community Council Directive of 24 November 1986 (86/609 EEC) for the Care and Use of Laboratory Animals.
Pharmacological selectivity assays
The activity of CR4056 was first evaluated in vitro in screening assays to assess the pharmacological selectivity relative to cell-surface receptors, ion channels, transport sites and enzymes by the use of standardized assay protocols as described in . The following additional tests were carried out in order to further investigate the results of aforementioned in vitro screening assays.
Binding to imidazoline I2 receptors
Whole brains minus cerebellum from male Wistar rats were homogenized in buffered sucrose and centrifuged at 1000 × g for 10 minutes at 4°C. Supernatants were pooled, centrifuged at 32000 × g for 20 minutes at 4°C and the pellets, suspended in assay buffer (50 mM Tris-HCl, 1 mM MgCl2, pH 7.4 at 4°C), were washed three times by repeated centrifugation as above, and then stored at −80°C until use. Prior to radioligand binding studies, membrane pellets were thawed and washed a further 4 times by re-suspension and repeated centrifugation in assay buffer to remove any possible endogenous inhibitors of binding. Aliquots of membrane suspension were incubated for 90 minutes at 25°C with 2.5 nM tritiated (2-benzofuranyl)-2-imidazoline ([3H]2-BFI) in the absence or presence of different concentrations of test compound. Test compound diluent dimethyl sulfoxide (DMSO) 0.5% final concentration was added to total binding samples; non-specific binding was determined in the presence of 10 μM 2-(4,5-dihydroimidazol-2-yl)quinoline hydrochloride (BU224).
Binding to MAO enzymes
Binding assays on monoamine oxidases in rat cerebral cortical membranes were conducted according to previously published papers.Citation21,Citation22 In summary, for the MAO-A binding assay, tritiated N-(2-aminoethyl)-5-(m-fluorophenyl)-4-thiazole carboxamide HCl ([3H]Ro 41-1049, 10 nM) was incubated in the absence or presence of test compound for 60 minutes at 37°C and non specific binding was determined in the presence of 1 μM clorgyline. For the MAO-B binding assay, incubation of [3H]Ro 19-6327(tritiated lazabemide) (15 nM) was carried out for 90 minutes at 22°C and non specific binding was determined in the presence of 10 μM (R)-deprenyl.
Human recombinant MAO functional assays
MAO-A and MAO-B enzymes were prepared from insect cells infected with recombinant baculovirus containing cDNA inserts for the human MAO genes. In these experiments, an aminopropylether analog of luciferin methyl ester was used as a substrate for the MAO reaction. In the first assay step, the enzyme oxidizes the substrate to form luciferin methyl ester. In the second step, the methyl ester is hydrolyzed and luciferin is oxidized to produce light. Luminescence was measured with a luminometer and displayed as relative light units (RLU). Half maximal inhibitory concentration (IC50) values were calculated by linear regression. Cumulative curves analysis (at least 2 independent experiments; log nM or log μM concentration of test compound versus % inhibition) was performed by using the ALLFIT program.
In vivo quantification of endogenous norepinephrine tissue levels
Male Wistar rats were orally administered with vehicle or CR4056 (20 mg/kg). Two hours later the animals were killed by decapitation, and brains or spinal cords were quickly removed and transferred in ice-cold artificial cerebrospinal fluid (aCSF) of the following composition (mM):125 NaCl, 3 KCl, 1.2 CaCl2, 1.2 MgSO4, 1 NaH2PO4, 22 NaHCO3, and 10 glucose (aerated with 95% O2 and 5% CO2 at 37°C), pH 7.2–7.4. Levels of endogenous norepinephrine (NE) were measured in parieto-occipital cortices, lumbar spinal cords (L4–L6), and plasma samples using a commercial enzymatic immunoassay kit (Noradrenaline Research EIA, Nordhorn, Germany).
In vivo models of experimental pain
CR4056 was evaluated in well characterized in vivo models to assess inflammatory and neuropathic pain. Unless otherwise noted, all experimental and control groups contained at least six animals per group. Statistical analysis was performed by one-way analysis of variance (ANOVA), with P < 0.05 accepted as significant. Inter-group differences were assessed by appropriate post hoc comparison tests. All data were expressed as the mean ± standard error of the mean (SEM).
Complete Freund’s adjuvant (CFA)-induced mechanical hyperalgesia
Unilateral inflammation was induced by injecting 100 μL of a 50% solution of CFA (Sigma Aldrich, Milan, Italy) in physiological saline into the plantar surface of the right hind paw of the rat. CFA was injected 24 hours before CR4056 administration. The rats were fasted overnight before experimental use. At the time of the experiment, animals were gently restrained, and steadily increasing pressure was applied to the dorsal surface of the CFA-treated paw via a dome-shaped plastic tip. Paw withdrawal threshold (PWT) to mechanical pressure was measured with a Randall-Selitto Analgesy-Meter (Ugo Basile, Comerio Varese, Italy).
Capsaicin-induced mechanical hyperalgesia
Rats were fasted overnight before drug administration. A first measurement of pain threshold (Randall–Selitto test) was undertaken before capsaicin injection. Capsaicin (Sigma Aldrich, Milan, Italy) was administered at time t = 0 by the intraplantar route in the right hind paw (10 μL of a 1 mg/mL Tween 80/saline solution). Sixty minutes later, animals were dosed by the oral route with CR4056 (3–30 mg/kg) or its vehicle in a volume of 5 mL/kg. A sham control group was always present for comparison. In mechanistic studies, antagonists were administered 30 minutes after capsaicin and 15 minutes before CR4056 administration.
Streptozotocin-induced neuropathic (diabetic, type I) pain in rats
Diabetes was induced in rats by administration of a single dose of streptozotocin (STZ; 75 mg/kg, i.p.). Twelve and twenty-four days after STZ injection, the presence of diabetes was confirmed by measuring blood glucose concentrations. Animals with values lower than 250 mg/dL were excluded from the studies. CR4056 was orally administered at doses of 6, 20, or 60 mg/kg four weeks after the STZ injection. Paw withdrawal threshold (PWT) to mechanical pressure was measured with a Randall–Selitto Analgesy-Meter (Ugo Basile, Comerio Varese, Italy).
Acid-induced muscle allodynia in rats
Male Wistar rats were brought to the behavioral testing room 1 hour before the test to acclimatize them to the testing environment. The right gastrocnemius muscle was injected with 150 μL of pH = 4 preservative-free sterile saline. Five days later (d5), the same gastrocnemius muscle was re-injected using an identical injection protocol. As a control for the injection procedure, a separate group of animals were injected with sterile saline. Ipsilateral and contralateral paw withdrawal thresholds were measured in response to mechanical stimuli on days 0 (baseline – 0d), 5 (5d) and 24 hours after the second acid injection (6d). Nociceptive thresholds, expressed in grams (g), were measured using a Dynamic Plantar Aesthesiometer (Ugo Basile, Comerio Varese, Italy) by applying increasing pressure to the right and left hind paw until the rat withdrew the paw.Citation23 A maximal cut-off of 50 g was used to prevent tissue damage. The threshold was tested three times for each paw and the mean value was calculated. CR4056 was administered as oral suspension at doses of 6, 20 and 60 mg/kg in comparison to intraperitoneal gabapentin (30 mg/kg) dissolved in saline. Mechanical withdrawal thresholds of both hind paws were measured 1 hour and 30 minutes after CR4056 and gabapentin injection respectively. One-way analysis of variance (ANOVA) followed by post hoc analysis for multiple comparisons were used to evaluate the differences between groups for each pharmacological treatment: a paired t-test was performed to evaluate the differences between the mechanical withdrawal thresholds before the first acidic saline injection (0d) and 24 hours after the second injection (6d), before pharmacological treatments.
Motor function assays
Locomotor activity was measured in an open field arena by using an overhead video camera coupled to a microcomputer by an image analyzer (EthoVision, Noldus Information Technology, Sterling, VA) used to track movement of rats in the arena. A remote switch was used to start and stop tracking. For the rotarod assay, rats were allowed a 30 minute acclimatization period in the testing room and then placed on a 9 cm diameter rod, which increased in speed from 0 to 20 rpm over a 60 second period. The time required for the rat to fall from the rod was recorded, with a maximum score of 60 seconds. Each rat was given three training sessions.
Chemicals
The following chemicals were used: capsaicin, naloxone, efaroxan, yohimbine, prazosin, idazoxan, streptozotocin, clonidine, clorgyline, (R)-deprenyl, human recombinant MAO-A and MAO-B (Sigma Aldrich, Milan, Italy); 2-(2-benzofuranyl)-2-imidazoline (2-BFI) and 2-(4,5-dihydroimidazol-2-yl)quinoline hydrochloride (BU-224) (Tocris Cookson, Bristol, UK). [3H]-2-BFI, [3H]Ro 41-1049 and [3H]Ro 19-6327 (GE Healthcare, Milan, Italy); MAO-Glo assay kit (Promega, Madison, WI). CR4056 (2-phenyl-6-(1H-imidazol-1yl)quinazoline) was synthesized by the Medicinal Chemistry Department of Rottapharm. For in vitro assays CR4056 stock solutions (10 mM) in DMSO were prepared, diluted in the corresponding assay buffer and used the same day. For in vivo oral administration, CR4056 was suspended in 0.5% methyl cellulose (MC) in 5 mL/kg injection volume; all the other drugs were dissolved in 0.9% normal saline for intraperitoneal (i.p.) or subcutaneous (s.c) administration.
Results
Receptor characterization panel
A receptor characterization panel including adrenergic, serotonergic, dopaminergic, opioid, cholinergic (muscarinic and nicotinic), and sigma receptors, as well as some amino acid receptor types (gamma-aminobutyric acid [GABA], glycine, glutamate) was performed. Percent inhibition of specific binding at the concentration tested (10 μM) is reported in . Moreover, a limited enzyme characterization panel including cycooxygenase-2 (COX2), inducible NO synthase (iNOS), TNF-α converting enzyme, carbonic anhydrase II, phosphoinositide 3-kinases (PI 3-Kβ, PI 3-Kγ, PI 3-Kδ), catechol-O-methyl transferase (COMT) and human mTOR kinase was performed (not shown). CR4056 was inactive in all assays but inhibited the affinity for I2 imidazoline receptors and monoamine oxidases A (MAO-A) by more than 90%.
Binding to imidazoline I2 receptors
The purpose of this study was to investigate the effects of CR4056 in a receptor binding assay selective for the I2 receptor subtype. We used rat whole-brain membranes labeled with the specific radioligand [3H]2-BFI. Matching experiments with various reference compounds were run for comparison. summarizes the effects of CR4056 and reference compounds. CR4056 inhibited [3H]2-BFI binding with an affinity value (IC50) of 596 ±76 nM (n = 3). Affinities of reference compounds were in line with published data.
Binding and functional activity assays on MAO
The effect of CR4056 was studied in receptor binding assays to evaluate its affinity for MAO-A and MAO-B sites in rat cerebral membranes. The refeDrence agents were tested concurrently with the test compound in order to assess the assay suitability. CR4056 inhibited MAO-A specific binding with an IC50 of 358 ± 12 nM (n = 3). Inhibition of MAO-B specific binding was achieved at concentrations two orders of magnitude higher. Binding assay results on rat cerebral native MAO and functional activity data, obtained on human recombinant enzymes, were shown to be consistent with each other. summarizes the effects of CR4056 and reference compounds on the activity of mono-amine oxidases. CR4056 selectively inhibited the enzymatic activity of MAO-A, with an IC50 value of 202.7 ± 10.3 nM (n = 3). As for the binding study, CR4056 was about 100 times less active as an inhibitor of MAO-B activity. The activity and selectivity of the reference compounds were in agreement with published data.
In vivo effects on endogenous norepinephrine (NE) levels
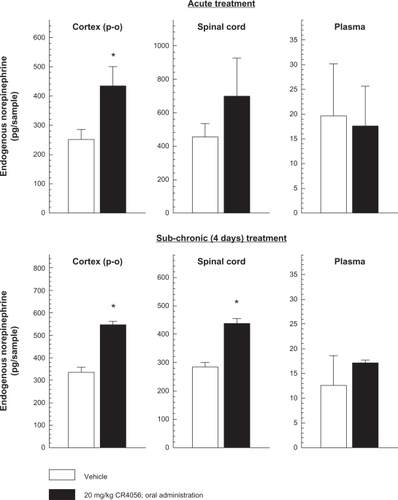
Two hours after a single oral dose of 20 mg/kg CR4056, endogenous NE levels increased by 68.2% ± 14.1% (P < 0.05 versus vehicle; Student’s t-test) in the parieto-occipital cortex. The effect of CR4056 at the spinal cord level was not statistically significant, but there was a clear tendency for NE levels to increase (). No effect was found in plasma samples. Sub-chronic (four days) oral treatment with 20 mg/kg CR4056 once daily, significantly increased NE levels both in the cerebral cortex (63.1% ± 4.2%; P < 0.05 versus vehicle; Student’s t-test) and in the lumbar spinal cord (51.3% ± 6.7%; P < 0.05 versus vehicle; Student’s t-test).
Figure 1 Effects of acute and sub-chronic oral treatment (once daily for 4 days) with CR4056 on endogenous norepinephrine (NE) levels in rat parieto-occipital cortex, lumbar spinal cord (L4–L6) and plasma.
CFA-induced inflammatory pain in rats
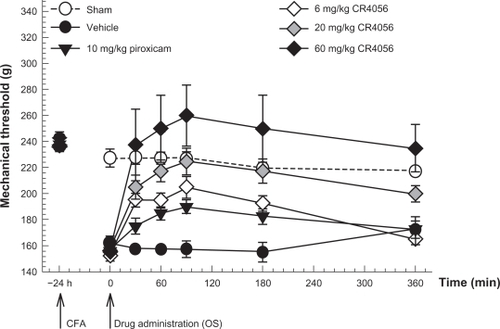
Male Wistar rats were injected into the right hind footpad with 50 μg of Mycobacterium tuberculosis in 100 μL vehicle (CFA). Twenty-four hours later, CR4056 (6, 20, or 60 mg/kg) or piroxicam (10 mg/kg) was administered by the oral route and the response to noxious mechanical stimulation was assessed by measuring the paw withdrawal threshold (PWT) (). In this experimental model of inflammatory pain, CR4056 dose-dependently reduced mechanical hyperalgesia (effective dose [ED50] = 5.8 mg/kg) (Kruskal–Wallis one way ANOVA: H[5, 36] = 27.23, P < 0.001). A complete reversion of CFA-induced hyperalgesia was achieved at the calculated ED100 dose of 22.3 mg/kg.
Figure 2 Antinociceptive effects of CR4056 on CFA-induced inflammatory pain in rats (Randall-Selitto test). CR4056 was orally administered 24 hours after a CFA injection in the right hind paw of the rats. Piroxicam (10 mg/kg; oral) was used as a positive control.
Capsaicin-induced neurogenic/inflammatory pain in rats
This experimental paradigm has been thoroughly investigated to understand the pharmacology of CR4056 analgesic properties. The effect of CR4056 on capsaicin-induced mechanical hyperalgesia was examined in male Wistar rats. Capsaicin (0.1%) or its vehicle (saline containing 1.5% Tween 80 and 1.5% ethanol) was injected subcutaneously into the plantar hind paw. The total injected volume was 10 μL. One hour after the challenge, animals received oral CR4056 (3, 10, or 30 mg/kg), piroxicam (10 mg/kg) or vehicle. Paw withdrawal threshold was measured 1 hour before capsaicin treatment, immediately before the administration of test compounds and 1 and 2 hours later.
Baseline withdrawal threshold to mechanical pressure was about 230 g. Sixty minutes after capsaicin injection, withdrawal threshold decreased to 162 ± 15 g (P < 0.01 versus baseline). CR4056 (10 mg/kg) and piroxicam (10 mg/kg) significantly reversed the decrease in withdrawal threshold caused by capsaicin (P < 0.001 versus vehicle; Bonferroni t-test) (). The highest tested dose of CR4056 (30 mg/kg) completely reversed the effect of capsaicin, increasing PWT to 239 ± 12 g (P < 0.001 versus vehicle; Bonferroni t-test) after 1 hour.
Figure 3 Capsaicin-induced neurogenic/inflammatory pain in rats: effect of increasing oral doses of CR4056 (Randall–Selitto test). CR4056 (range: 3–30 mg/kg) dose-dependently reversed the mechanical hyperalgesia induced by an intraplantar injection of capsaicin (F[5, 36] = 27.57, P < 0.001). Piroxicam (10 mg/kg; oral) was used as a positive control.
![Figure 3 Capsaicin-induced neurogenic/inflammatory pain in rats: effect of increasing oral doses of CR4056 (Randall–Selitto test). CR4056 (range: 3–30 mg/kg) dose-dependently reversed the mechanical hyperalgesia induced by an intraplantar injection of capsaicin (F[5, 36] = 27.57, P < 0.001). Piroxicam (10 mg/kg; oral) was used as a positive control.](/cms/asset/3969cfd3-6827-45c4-b20f-7139cf993c12/djpr_a_18353_f0003_b.jpg)
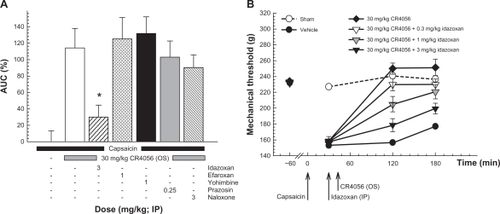
In vitro data clearly demonstrated a selective interaction of CR4056 with imidazoline I2 receptors. We therefore tried to better characterize the analgesic effect of the compound by administering different antagonists of imidazoline and/or adrenergic receptors to capsaicin-treated rats 15 minutes before a full analgesic dose of CR4056 (30 mg/kg). Moreover, because high doses of CR4056 could mimic the effect of some opioid agonists (ie, morphine) and cause hypoalgesia, we also tested the hypothesis of an indirect in vivo modulation of the opioidergic system by CR4056. For this purpose, capsaicin-treated rats were given naloxone, a μ-opioid antagonist, 15 minutes before CR4056 administration. Efaroxan (a non-selective antagonist of imidazoline I1 and adrenergic α2 receptors), yohimbine (a selective α2 antagonist) and prazosin (a selective α1 antagonist) did not influence the effect of CR4056 on capsaicin-induced mechanical hyperalgesia (, Panel A). Conversely, idazoxan (3 mg/kg; a non-selective antagonist of imidazoline I2 and adrenergic α2 receptors) greatly reduced the analgesic action of CR4056 (74.1% ± 12.7%, P < 0.001 versus group of animals treated with 30 mg/kg CR4056 only). Naloxone antagonized the slight hypoalgesic effect previously reported but not the analgesic effect induced by CR4056. In a confirmatory experiment, idazoxan (0.3, 1, and 3 mg/kg i.p.) dose-dependently inhibited the analgesic effect of CR4056 by decreasing PWT (increased hyperalgesia) in capsaicin-treated rats (, Panel B) (Kruskal–Wallis one way ANOVA: H [5, 71] = 48.50, P < 0.001). The maximal inhibition observed in the presence of 3 mg/kg idazoxan was 76.1% ± 9.5%, consistent with the previous study.
Figure 4 Capsaicin-induced neurogenic/inflammatory pain in rats (Randall–Selitto test). Panel A: effects of different receptor antagonists on the analgesic activity induced by 30 mg/kg oral CR4056.
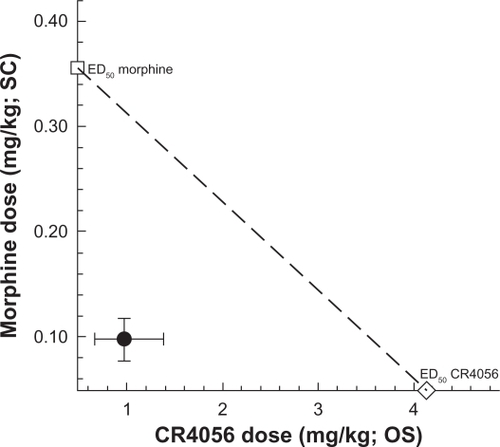
We also examined the effects of co-administered morphine and CR4056 and determined the type of interaction between the two treatments. Morphine (0.03–3 mg/kg, subcutaneously), CR4056 (0.3–30 mg/kg, orally) and their combination produced a significant, dose-dependent anti-hyperalgesic effect in capsaicin-injected rats. Isobolographic analysis revealed a significant synergistic interaction between morphine and CR4056 (). When these agents were combined, the doses needed to reach the median effective dose were about 4-fold lower than those seen after administration of each drug alone (ED50: 0.98 versus 4.13 mg/kg for CR4056, 0.097 versus 0.36 mg/kg for morphine).
Figure 5 Isobologram for the effects of CR4056 and morphine, alone or in combination, in capsaicin-induced neurogenic/inflammatory pain in rats. Filled circle corresponds to the experimental co-treatment ED50 with 95% confidence limits; open square corresponds to the experimental ED50 for morphine alone, and open diamond corresponds to the experimental ED50 for CR4056 alone.
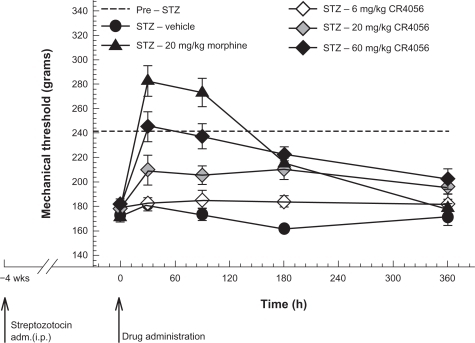
Streptozotocin-induced neuropathic (diabetic, type I) pain in rats
CR4056 dose-dependently decreased STZ-induced diabetic pain in rats (F[4, 35] = 31.27, P < 0.001) (). Oral treatment at a dose of 60 mg/kg completely reversed mechanical hyperalgesia within 30 minutes. Moreover, AUC analysis (0–6 hours) showed that 60 mg/kg CR4056 decreased by 84% the total STZ-induced hyperalgesic effect (P < 0.001 versus vehicle, Bonferroni t-test). The μ-opioid receptor agonist morphine (20 mg/kg, s.c.) completely reversed the hyperalgesia at 30 and 90 minutes after treatment, with some degree of hypoalgesia. However, PWT at 6 hours in morphine-treated rats was not different from that in untreated diabetic rats. Conversely, the analgesic effect of CR4056 lasted longer: a 60 mg/kg dose significantly reduced mechanical hyperalgesia by 58% at 6 hours post-dosing (P < 0.05 versus vehicle, Bonferroni t-test).
Figure 6 Streptozotocin (STZ)-induced neuropathic (diabetic, type I) pain in rats: effects of increasing oral doses of CR4056 (Randall–Selitto test). Diabetes was induced in rats by administration of a single dose of STZ (i.p.). CR4056 was orally administered four weeks after the STZ injection. Morphine (20 mg/kg; s.c.), was used as a positive control.
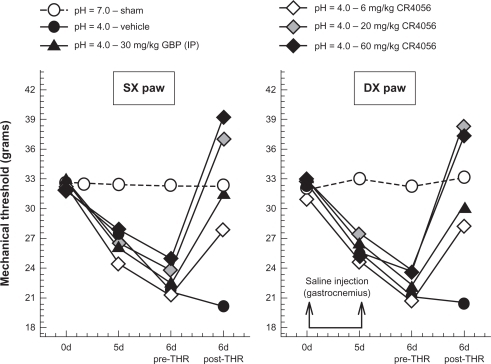
Acid-induced muscle allodynia in rats
The acidic saline animal model of pain is thought to mimic human chronic pain syndromes such as fibromyalgia. Repeated intramuscular injections of acidic saline is a model of non-inflammatory pain characterized by bilateral long-lasting allodynia of the paw, which is believed to be centrally mediated.Citation24
CR4056 significantly increased the mechanical withdrawal thresholds of both ipsilateral (F[4, 30] = 19.97, P < 0.001) and contralateral (F[4, 30] = 31.58, P < 0.001) hind paws compared with vehicle control (). A pairwise multiple comparison procedure (Holm–Sidak method) further evidenced that the anti-nociceptive effects of 6 mg/kg CR4056 and 30 mg/kg gabapentin were similar. CR4056 20 and 60 mg/kg completely reversed acidic saline induced allodynia. No statistically significant difference was detected between the groups treated with CR4056 at 20 and 60 mg/kg.
Figure 7 Antiallodynic effects of CR4056 in the acid-induced muscle allodynia model in rats (Dynamic Plantar Aesthesiometer; Ugo Basile, VA, Italy). Right gastrocnemius muscle was injected with acidic saline (pH = 4). Five days later (d5), the same gastrocnemius muscle was re-injected using an identical injection protocol. As a control for the injection procedure, a separate group of animals were injected with sterile saline. Six days after the first acidic saline injection, CR4056 was orally administered to rats two hours before testing. Gabapentin (GBP) (30 mg/kg; i.p.) was used as positive control.
Abbreviations: DX, right paw; SX, left paw.
Motor function: rotarod and open field tests
International requirements for central nervous system (CNS) safety pharmacology include assessment of motor coordination and locomotor activity. The rotarod test provides an estimation of the animal’s level of neuromuscular coordination.
Oral administration of CR4056 in a dose range of 3–30 mg/kg did not cause a deterioration of animals’ performance in a rotarod test. In addition, an open field test was used to measure locomotor activity. There were no statistical differences in locomotor activity levels after CR4056 treatment (3–30 mg/kg by oral administration) one hour after administration. Indeed, we repeated the test six hours after CR4056 administration and after four days of treatment (once daily) without any noticeable differences in the motor behavioral pattern.
Discussion
CR4056 is a new ligand of the imidazoline-2 sites (I2R) with efficacy in several models of pain. In particular CR4056, administered by the oral route, almost invariably showed a complete reversion of hyperalgesia induced by local (CFA) or neurogenic (capsaicin) inflammation, and by experimental neuropathies (diabetes and acid-induced muscle allodynia). In all of these paradigms, doses of CR4056 below 20 mg/kg were highly effective against the exaggerated pain sensation, while higher doses proved to be, as in the case of opiates, decidedly hypoalgesic.
However, as shown in vitro by a broad binding screening panel, CR4056 does not interact directly with any of the known opioid receptors. In the last few years Cheng et alCitation25,Citation26 significantly contributed to the explanation as to why a modulator of imidazoline-2 receptors should mimic the effect of some opioid receptor ligands. In fact, they showed that agmatine, the endogenous ligand of imidazoline receptors, increased beta-endorphin secretion in the rat adrenal medulla and this effect was blocked by the I2R putative antagonists BU-224 and amiloride. This peripheral beta-endorphin increase will possibly be additive to the well described central effect induced by I2 ligandsCitation27 on the firing rate of locus coeruleus neurons, where there is a cross-link between I2 sites and the opioid system. This interaction leads to an attenuation of both the hyperactivity of locus coeruleus neurons during opiate withdrawal and the development of tolerance to morphine when agmatine or 2-BFI is chronically administered.Citation18
In this paper, the pharmacology of the analgesic activity of CR4056 was examined in vivo using the simple model of capsaicin-induced neurogenic pain. In this model, it has been clearly demonstrated that, while the concomitant treatment with α1, α2, or I1 antagonists did not affect the efficacy of CR4056, treatment with the nonselective I2 antagonist idazoxan dose-dependently abrogated the effect of a fully active dose of CR4056, suggesting that the binding with the I2 site is a primary requirement for its efficacy. Interestingly, naloxone, an opiate antagonist, was only able to dampen the hypoalgesic effect of CR4056, but not its analgesic properties, suggesting that the hypoalgesic component could be due to the release of endogenous endorphins, as observed with other I2 ligands.Citation26
In this respect, the capsaicin model was also useful to evaluate a possible synergy between morphine and CR4056. Morphine is the most commonly used opioid for the treatment of severe pain, but adverse effects, including nausea and respiratory depression, limit its use. Combination therapy is a valid approach in pain management because optimal analgesia can be reached at lower doses of each drug than those required in monotherapy. Generally, if different mechanisms jointly contribute to pain control, a synergistic interaction is considered likely. Such combination may decrease the necessary dose of individual drugs, thereby increasing the maximum analgesic effect with a decreased incidence of adverse effects. Conversely, if the mechanisms of action of one drug are involved in those of another drug, a synergistic interaction is not expected. The combination of CR4056 with morphine has been found to be clearly synergic, being about three- to four-fold more effective than predicted on the basis of a simple additive effect.
Potential interactions with other pain modulatory systems, such as the cannabinoidCitation28,Citation29 pathway, have been proposed in the last few years to explain the efficacy of imidazoline site ligands in preclinical models. For instance, Aggarwal et alCitation29 found a synergistic effect of agmatine and WIN 55212-2 (a CB1 cannabinoid agonist) in the hot-plate assay of thermal nociception in rats. They also found that the mixed I2/α2 antagonist idazoxan, but not the selective α2 antagonist yohimbine, dose-dependently antagonizes the enhanced response-latency produced by co-administration of agmatine and WIN 55212-2. In exploratory experiments we tested whether a commercially available CB1 antagonist (AM-251) could antagonize the analgesic effect of CR4056 in the rat model of capsaicin-induced neurogenic pain. Unlike idazoxan, AM-251 had no effect under the experimental condition chosen (data not shown), suggesting that the cannabinoid pathway is not involved in the mechanisms underlying CR4056-induced analgesia.
Another important issue to consider in the evaluation of CR4056 mechanism of action is its effect on monoamine oxidase A. Although in the literature there is still no consensus about the nature of I2-MAO interaction,Citation6,Citation7,Citation30 most I2R ligands are reported to inhibit the activity of MAO, thereby inducing a net increase of endogenous catecholamines available at synaptic level. CR4056 was found to selectively inhibit the activity of human recombinant MAO-A with sub-micromolar affinity and, when administered orally to rats, to increase the norepinephrine tissue levels both in the cerebral cortex and in the lumbar spinal cord. The role of norepinephrine in controlling spinal and supraspinal pain modulatory effects is well documented by both preclinical and clinical data.Citation31–Citation34 In particular, as a general mechanism of the noradrenergic theory of pain, peripheral nociceptive stimuli induce the activation of noradrenergic projections from higher centres including the locus coeruleus to the spinal cord, resulting in antinociception and inhibition of spinal nociceptive neurons. This story becomes complicated due to some findings showing that noradrenergic neurons of the locus coeruleus do not always induce pain inhibition, and can also generate paradoxical facilitation, probably mediated by synaptic afferents to the rostral ventral medulla (RVM), a well known pain facilitatory centre.Citation35–Citation37 However, current therapeutic guidelines clearly state that drugs able to enhance the central synaptic levels of norepinephrine (ie, TCAs and/or the newer mixed norepinephrine/serotonin re-uptake inhibitors –[SNRIs]) are one of the first line treatments recommended for the management of neuropathic pain conditions.Citation20,Citation38,Citation39 Moreover, recent findings show that metabolism of catecholamines by sensory neurons contributes to peripheral neuropathies by producing neurotoxic metabolites, suggesting that MAO-A inhibitors devoid of central adverse effects could be useful for the treatment of neuropathic pain.Citation40 This could be the case for functional MAO-A inhibitors, such as CR4056, modulating the enzyme activity through accessory sites present in discrete populations of enzymes.Citation4,Citation5
During the pre-clinical development path enabling CR4056 to enter into human clinical studies, safety pharmacology findings were consistent with the activity of CR4056 on the catecholamine system. At supra-pharmacological doses in the conscious dog, CR4056 induced slight hypertensive effects associated with tachycardia. Consistently, tachypnea associated with a respiratory stimulant effect was observed in the conscious rat. Interestingly, all these effects on cardio-respiratory functions can be produced by excitation of afferent cardiac sympathetic nerve fibers and are functionally opposite to what is generally observed after administration of analgesic opiates. It is well known that morphine administration in rats and dogs decreases heart rate, blood pressure, and cardiac output.Citation41 Additionally, all opiates acting at the μ receptor induce a marked respiratory depression that complicates their clinical administration and that is potentially life threatening in the case of opiate overdose.Citation42 This could be relevant in view of the demonstrated synergy between morphine and CR4056. On the other hand, the absence of respiratory depression, hypotension, and bradycardia in animals treated with CR4056 rules out the possibility that this compound directly activates central α2 receptors, whose stimulation by drugs like clonidine, in case of overdosage, induces hypotension, bradycardia, sedation, miosis, and respiratory depression.Citation43
Besides the above described effects, when tested in the pharmacological/toxicological range in rats, CR4056 induced a dose-dependent diuretic effect. Notably, the diuretic effect of CR4056 is shared by several imidazoline receptor ligands such as the diuretic amiloride, a putative selective ligand for the I2A subtype,Citation44,Citation45 and the anti-hypertensive drug moxonidine, which is considered to be selective for imidazoline I1 receptors even though its diuretic activity is blocked by the I2/α2 antagonist idazoxan.Citation46
Conclusion
CR4056 is a very effective analgesic compound active in several preclinical models relevant for important human pathologies including fibromyalgia and diabetes-induced neuropathy. CR4056 has now completed preclinical development and a phase I safety study in humans has now been designed to finally develop the compound as a first in class I2 ligand in chronic and neuropathic pain conditions.
Acknowledgements
We are indebted to Professor Jesús Andrés García Sevilla for a helpful discussion on the role of I2 receptors, and to Dr Michael Pilkington-Miksa for his kind English revision of the manuscript. The authors wish to thank Mr Dario Tremolada, Mr Luca Catapano, Mrs Anna Stucchi, Mrs Raffaella Miotto for their technical support, Mrs Laura Radaelli for her skillful secretarial assistance, and Dr Albino Bonazzi for his painstaking piece of institutional reporting on this project.
Supplementary tables
Table S1 CR4056 receptor characterization panel
Table S2 Effects of CR4056 and reference compounds on [3H]2-BFI binding to the imidazoline I2 receptor in rat cerebral membranes
Table S3 Effects of CR4056 and reference compounds on the enzymatic activity of human recombinant MAO-A and MAO-B
Table S4 Acid-induced muscle allodynia in rats
Disclosure
This study was funded by the Rottapharm group. However, the Rottapharm group as a corporate entity had no role in the conduct of the study; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication.
Preliminary results of this study were presented in abstract form at the 6th Forum of European Neuroscience (Lanza et al, FENS Abstr., vol.4, 189.18, 2008), held in Geneva, Switzerland; at the Keystone Symposia “The Neurobiology of Pain and Analgesia” (Lanza et al, Keystone Abstr., C2.2.201, 2009), held in Santa Fè, New Mexico, USA; at the 13th National Congress of the Italian Society for Neuroscience (Neri et al, SINS Abstr., P-64, 2009), held in Milan, Italy; and at the 3rd International Congress on Neuropathic Pain (Chiorazzi et al, European Journal of Pain Supplements, 4(1), 55, 2010) held in Athens, Greece.
References
- MolderingsGJImidazoline receptors: basic knowledge, recent advances, and future prospects for therapy and diagnosisDrug Future199722757772
- PiletzJEIvanovTRSharpJDImidazoline receptor antisera-selected (IRAS) cDNA: cloning and characterizationDNA Cell Biol20001931932910882231
- BousquetPIdentification and characterization of I1 imidazoline receptors: their role in blood pressure regulationAm J Hypertens20001384S88S10921526
- TessonFLimon-BoulezIUrbanPLocalization of I2-imidazoline binding sites on monoamine oxidasesJ Biol Chem1995270985698617730367
- CarpénéCCollonPRemauryAInhibition of amine oxidase activity by derivatives that recognize imidazoline I2 sitesJ Pharmacol Exp Ther19952726816887853182
- OzaitaAOlmosGBoronatMALizcanoJMUnzetaMGarcía-SevillaJAInhibition of monoamine oxidase A and B activities by imidazol(ine)/guanidine drugs, nature of the interaction and distinction from I2-imidazoline receptors in rat liverBr J Pharmacol19971219019129222546
- McDonaldGROlivieriARamsayRRHoltAOn the formation and nature of the imidazoline I2 binding site on human monoamine oxidase-BPharmacol Res201062647548820832472
- KimuraATyackeRJRobinsonJJIdentification of an imidazoline binding protein: creatine kinase and an imidazoline-2 binding siteBrain Res20091279212819410564
- HoltASmithDJCendronLZanottiGRigoADi PaoloMLMultiple binding sites for substrates and modulators of semicarbazide-sensitive amine oxidases: kinetic consequencesMol Pharmacol20087352553817989349
- García-SevillaJAEscribáPVSastreMImmunodetection and quantitation of imidazoline receptor proteins in platelets of patients with major depression and in brains of suicide victimsArch Gen Psychiatry1996538038108792757
- García-SevillaJAEscribáPVWalzerCBourasCGuimónJImidazoline receptor proteins in brains of patients with Alzheimer’s diseaseNeurosci Lett199824795989655601
- PiletzJEZhuHOrdwayGImidazoline receptor proteins are decreased in the hippocampus of individuals with major depressionBiol Psychiatry20004891091911074229
- BoronatMAOlmosGGarcía-SevillaJAAttenuation of tolerance to opioid-induced antinociception and protection against morphine-induced decrease of neurofilament proteins by idazoxan and other I2-imidazoline ligandsBr J Pharmacol19981251751859776358
- FairbanksCASchreiberKLBrewerKLAgmatine reverses pain induced by inflammation, neuropathy, and spinal cord injuryProc Natl Acad Sci200097105841058910984543
- AriciogluFKorcegezEBozkurtAOzyalcinSEffect of agmatine on acute and mononeuropathic painAnn N Y Acad Sci2003100910611515028574
- GentiliFCardinalettiCCarrieriAInvolvement of I2-imidazoline binding sites in positive and negative morphine analgesia modulatory effectsEur J Pharmacol2006553738117081513
- WuNSuRBLiJAgmatine and imidazoline receptors: their role in opioid analgesia, tolerance and dependenceCell Mol Neurobiol20082862964117653850
- Ruiz-DurantezETorrecillaMPinedaJUgedoLAttenuation of acute and chronic effects of morphine by the imidazoline receptor ligand 2-(2-benzofuranyl)-2-imidazoline in rat locus coeruleus neuronsBr J Pharmacol200313849450012569074
- BennettMIEffectiveness of antiepileptic or antidepressant drugs when added to opioids for cancer pain: systematic reviewPalliat Med201010.1177/0269216310378546
- FinnerupNBSindrupSHJensenTSThe evidence for pharmacological treatment of neuropathic painPain201015057358120705215
- CesuraAMGalvaMDImhofRKyburzEPicottiGBDaPradaM[3H]Ro 19-6327: a reversible ligand and affinity labelling probe for monoamine oxidase-BEur J Pharmacol19891624574652744079
- CesuraAMBosMGalvaMDImhofRDaPradaMCharacterization of the binding of [3H]Ro 41-1049 to active site of human monoamine oxidase-AMol Pharmacol1990373583662314388
- KimSHSongJMunHParkKUEffect of the combined use of tramadol and milnacipran on pain threshold in an animal model of fibromyalgiaKorean J Intern Med20092413914219543493
- NielsenANMathiesenCBlackburn-MunroGPharmacological characterisation of acid-induced muscle allodynia in ratsEur J Pharmacol20044879310315033380
- HwangSLLiuIMTzengTFChengJTActivation of imidazoline receptors in adrenal gland to lower plasma glucose in streptozotocin-induced diabetic ratsDiabetologia20054876777515756537
- ChangCHWuHTChengKCLinHJChengJTIncrease of beta-endorphin secretion by agmatine is induced by activation of imidazoline I(2A) receptors in adrenal gland of ratsNeurosci Lett201046829729919913596
- Ruiz-DurántezERuiz-OrtegaJAPinedaJUgedoLEffect of agmatine on locus coeruleus neuron activity: possible involvement of nitric oxideBr J Pharmacol20021351152115811877321
- RawlsSMTallaridaRJZiskJAgmatine and a cannabinoid agonist, WIN 55212-2, interact to produce a hypothermic synergyEur J Pharmacol2006553899817109846
- AggarwalSShavalianBKimERawlsSMAgmatine enhances cannabinoid action in the hot-plate assay of thermal nociceptionPharmacol Biochem Behav20099342643219538988
- MacInnesNHandleySLRegion-dependent effects of acute and chronic tranylcypromine in vivo on [3H]2-BFI binding to brain imidazoline I2 sitesEur J Pharmacol200142822122511675039
- JasminLTienDJanniGOharaPTIs noradrenaline a significant factor in the analgesic effect of antidepressants?Pain20031063814581104
- PertovaaraANoradrenergic pain modulationProg Neurobiol200680538317030082
- FinnerupNBA review of central neuropathic pain statesCurr Opin Anaesthesiol20082158658918784483
- BannisterKBeeLADickensonAHPreclinical and early clinical investigations related to monoaminergic pain modulationNeurotherapeutics2009670371219789074
- OssipovMHLaiJMalanTPPorrecaFSpinal and supraspinal mechanisms of neuropathic painAnn N Y Acad Sci2000909122410911921
- DubnerRThe neurobiology of persistent pain and its clinical implicationsSuppl Clin Neurophysiol2004573716106600
- BrightwellJJTaylorBKNoradrenergic neurons in the locus coeruleus contribute to neuropathic painNeuroscience200916017418519223010
- DworkinRHO’ConnorABBackonjaMPharmacologic management of neuropathic pain: evidence-based recommendationsPain200713223725117920770
- MarksDMShahMJPatkarAAMasandPSParkG-YPaeC-USerotonin-norepinephrine reuptake inhibitors for pain control: premise and promiseCurr Neuropharmacol2009733133620514212
- DinaOAKhasarSGAlessandri-HaberNNeurotoxic catecholamine metabolite in nociceptors contributes to painful peripheral neuropathyEur J Neurosci2008281180119018783367
- NapierLDStanfillAYoshishigeDAJacksonKEBarronBACaffreyJLAutonomic control of heart rate in dogs treated chronically with morphineAm J Physiol1998275H2199H22109843820
- McCrimmonDRAlheidGFOn the opiate trail of respiratory depressionAm J Physiol Regul Integr Comp Physiol2003285R1274R127514615398
- Klein-SchwartzWTrends and toxic effects from pediatric clonidine exposuresArch Pediatr Adolesc Med200215639239611929375
- SweetmanSMartindale: The Complete Drug Reference36th edLondonPharmaceutical Press2009
- OlmosGAlemanyRGarcía-SevillaJAPharmacological and molecular discrimination of brain I2-imidazoline receptor subtypesNaunyn Schmiedebergs Arch Pharmacol19963547097168971730
- SchlatterEAnkorina-StarkIHaxelmansSHohageHMoxonidine inhibits Na+/H+ exchange in proximal tubule cells and cortical collecting ductKidney Int1997524544599264001