Abstract
Although bortezomib (BTZ) is the frontline treatment for multiple myeloma, its clinical use is limited by the occurrence of painful peripheral neuropathy, whose treatment is still an unmet clinical need. Previous studies have shown chronic BTZ administration (0.20 mg/kg intravenously three times a week for 8 weeks) to female Wistar rats induced a peripheral neuropathy similar to that observed in humans. In this animal model of BTZ-induced neurotoxicity, the present authors evaluated the efficacy of CR4056, a novel I2 ligand endowed with a remarkable efficacy in several animal pain models. CR4056 was administered in a wide range of doses (0.6–60 mg/kg by gavage every day for 2–3 weeks) in comparison with buprenorphine (Bupre) (28.8 μg/kg subcutaneously every day for 2 weeks) and gabapentin (Gaba) (100 mg/kg by gavage every day for 3 weeks). Chronic administration of BTZ reduced nerve conduction velocity and induced allodynia. CR4056, Bupre, or Gaba did not affect the impaired nerve conduction velocity. Conversely, CR4056 dose-dependently reversed BTZ-induced allodynia (minimum effective dose 0.6 mg/kg). The optimal dose found, 6 mg/kg, provided a constant pain relief throughout the treatment period and without rebound after suspension, being effective when coadministered with BTZ, starting before or after allodynia was established, or when administered alone after BTZ cessation. A certain degree of tolerance was seen after 7 days of administration, but only at the highest doses (20 and 60 mg/kg). Bupre was effective only acutely, since tolerance was evident from the fourth day onwards. Gaba showed a significant activity only at the fourth day of treatment. CR4056, over the range of concentrations of 3–30 μM, was unable to hinder BTZ cytotoxicity on several tumor cell lines, which could indicate that this substance does not directly interfere with BTZ antitumor activity. Therefore, CR4056 could represent a new treatment option for BTZ-induced neuropathic pain.
Introduction
Peripheral neuropathy is a major dose-limiting side effect associated with anticancer chemotherapy.Citation1–Citation3 It is generally characterized by degeneration of sensory axons associated with chronic neuropathic pain (NP).Citation4 NP is a multifactorial condition resulting from damage or dysfunction of the nervous system that combines sensory loss in the painful neuropathic area and hypersensitivity phenomena such as hyperalgesia and allodynia.Citation5,Citation6 This side effect is also a dose-limiting toxicity for the new antineoplastic drug bortezomib (BTZ) (Velcade®).
BTZ is used for the treatment of refractory multiple myeloma (MM),Citation7,Citation8 and it is tested as an alternative drug for the treatment also of different hematological and solid neoplasms.Citation9,Citation10 The antineoplastic activity of BTZ is based mainly on the reversible inhibition of the 26S proteasome and nuclear factor–kappa B pathway signaling, sensitizing tumor cells to apoptosis.Citation11,Citation12 Hypersensitivity to sensory stimuli is a common source of NP in patients chronically administered with BTZ, even after treatment.Citation13
There are no drugs truly able to prevent or treat the painful symptoms associated with BTZ treatment. Radioligand binding and photoaffinity labeling studies have indicated that imidazoline binding sites represent a heterogeneous family of proteins that are currently grouped in three subtypes: I1, I2, and I3.Citation14–Citation16 In particular, the I2 subtype is expressed in brain regions important in mediating the stress response in animals.Citation17 Two decades of study on imidazoline binding sites evidenced their involvement in very different events such as cell proliferation, regulation of body fat, neuroprotection, inflammation, and pain.Citation16,Citation18 Although I2-like receptors are not fully characterized at the molecular level, robust pharmacological evidence has demonstrated an allosteric inhibition of monoamine oxidases (MAOs) enzymes by I2 ligands.Citation14,Citation19 Being responsible for monoamine catabolism, MAOs play a key role in the control of NP.Citation20,Citation21 Therefore, compounds able to interact with the I2 binding sites could represent a promising new class of drugs for use in the control of NP such as the painful peripheral neuropathy induced by BTZ.
CR4056 is a new synthetic compound () that interacts competitively with I2 binding sites and which inhibits allosterically the activity of human recombinant MAO-A. CR4056 has been proven to efficiently control the experimental pain in a variety of animal models.Citation22 Indeed, CR4056 in the dose range of 6–60 mg/kg was able to completely reverse in a dose-related manner the chronic inflammatory pain induced by complete Freund’s adjuvant in rats, as well as the acute neurogenic pain induced by capsaicin. Moreover, in the same dose range the compound was highly effective in two models of NP (ie, diabetic neuropathy and acid-induced fibromyalgia in rats) as described by Ferrari et al.Citation22 CR4056 is devoid of any affinity toward opiate receptors, so a direct involvement of these receptors could be excluded. However, it is known that activation of imidazoline I2 receptors in the adrenal gland induces beta-endorphin secretion and further activates mu opioid receptors in streptozotocin-diabetic rats.Citation23,Citation24 Therefore, an indirect involvement of endogenous opiates in the CR4056 hypoalgesic effect seen in several modelsCitation22 could not be completely ruled out. However, the antinociceptive activity of CR4056 has never been studied in a model of chemotherapy-induced NP. The aim of the present study is to investigate the potential antinociceptive effect of the I2 ligand CR4056 in a rat model of BTZ-induced NP.
Material and methods
Drugs
BTZ was from LC Laboratories (Woburn, MA), buprenorphine (Bupre) (Temgesic® 0.3 mg/mL injectable solution) was from Schering-Plough SpA (Milan, Italy), and CR4056 and Gaba were provided by Rottapharm | Madaus (Monza, Italy).
For in vitro studies BTZ and CR4056 were dissolved in dimethyl sulfoxide (DMSO) and diluted in culture medium before use. For in vivo experiments BTZ was dissolved in 5% polysorbate 80, 5% ethanol, and 90% sterile saline and was administered intravenously via the tail vein; CR4056 was dissolved in 0.5% methylcellulose and administered by oral gavage; Gaba and Bupre were both dissolved in sterile water and administered orally or subcutaneously, respectively.
In vivo studies
Animal husbandry
Female Wistar rats (Harlan Italy, Correzzana, Italy), each weighing 175–200 g on arrival at the housing room, were used. Animal husbandry followed the University of Milan Bicocca guidelines in compliance with national (Italian Legislative Decree 116/92, Gazzetta Ufficiale della Repubblica Italiana, No 40, February 18, 1992) and international laws and policies (European Economic Community Council Directive 86/609, OJ L 358, 1, December 12, 1987; US National Research Council’s Guide for the Care and Use of Laboratory Animals, 2011). The experimental plan was preliminarily examined and approved by the ethical committee of the University of Milan Bicocca (approval ID: 0015989). The rats were randomized and then housed two or three per cage in a limited-access animal facility, where room temperature and relative humidity were set at 22°C ± 2°C and 55% ± 10%, respectively. Artificial lighting provided a 24-hour cycle of 12 hours of light (7 am–7 pm) followed by 12 hours of darkness. The general condition of the rats was assessed daily and body weight was registered twice weekly.
Selection of doses
BTZ, Bupre, and Gaba doses were selected based on literature data.Citation25–Citation27 CR4056 doses were selected on the basis of the previous experience with this compound in other models of pain as published by Ferrari et al.Citation22 In particular, for the first experiment in bortezomib-induced neuropathy (Experiment 1) the present authors selected the same range of doses (6, 20, and 60 mg/kg) that proved to be active in two other models of NP in the rat – namely, the diabetes-induced neuropathy and the acid-induced fibromyalgia-like pain. In Experiment 2, considering the outstanding efficacy already shown after the low dose of 6 mg/kg, the range of doses was lowered (0.6, 2, and 6 mg/kg) to detect the minimal effective dose. In Experiment 3, only the optimal 6 mg/kg dose was tested in coadministration with BTZ.
Experiment 1
The experiment was planned as two consecutive phases, with phase 1 lasting 8 weeks and phase 2 lasting 2 weeks. At the beginning of the study, 70 rats were randomly divided into two groups: one group was treated intravenously with BTZ 0.2 mg/kg three times a week for 8 weeks (BTZ group; n = 50) and the other group was left untreated (control group [CTRL]; n = 20). After 8 weeks of treatment, at the beginning of phase 2, BTZ treatment was withdrawn and the rats were re-randomized to generate homogeneous groups on the basis of the results obtained from the dynamic test (see “Behavioral tests” section later in article). CTRL rats were allocated to one untreated group (CTRL; n = 10) and to one group orally treated with CR4056 at 60 mg/kg/day for 2 weeks (CR4056 60; n = 10). The BTZ-treated group was divided into five subgroups: the first one was left untreated but was manipulated exactly as the other rats (BTZ-fake-manipulated but untreated group [BTZ-fu]; n = 7), three groups received CR4056 6, 20, or 60 mg/kg/day for 2 weeks (BTZ + CR4056 6; BTZ + CR4056 20; BTZ + CR4056 60; n = 10 for each group), and the last group was treated subcutaneously for 2 weeks with Bupre at 28.8 μg/kg/day (BTZ + Bupre; n = 10).
Experiment 2
The experiment was planned as two consecutive phases, with phase 1 lasting 8 weeks and phase 2 lasting 3 weeks. At the beginning of the study, 72 rats were randomly divided into two groups: one group was treated with BTZ 0.2 mg/kg three times a week for 8 weeks (BTZ group; n = 60) and the other group was left untreated (CTRL; n = 12). After 8 weeks of treatment, at the beginning of phase 2, BTZ treatment was withdrawn and CTRL rats were left untreated; BTZ-treated rats were re-randomized on the basis of the results obtained from the dynamic test into five groups: the first group, as before, was left untreated (BTZ-fu; n = 12), three groups were treated orally with 0.6, 2, or 6 mg/kg/day of CR4056 for 3 weeks (BTZ + CR4056 0.6; BTZ + CR4056 2; BTZ + CR4056 6; n = 12 for each group), and the last group was treated orally with Gaba at 100 mg/kg/day for 3 weeks (BTZ + Gaba; n = 12). At the end of the 3-week period of treatment the rats were observed for an additional 1-week treatment-free period to evaluate possible withdrawal and/or rebound effects. The dynamic test was repeated at the end of this period also.
Experiment 3
The third experiment, planned as two consecutive phases – with phase 1 lasting 6 weeks and phase 2 lasting 4 weeks – was conducted to understand whether the antiallodynic effects could be confirmed in a coadministration protocol. At the beginning of the study, 51 rats were randomly divided into four groups: one group was left untreated (CTRL; n = 9); a second group was treated with CR4056 6 mg/kg/day orally (CR4056 group; n = 9); a third group was treated with BTZ 0.2 mg/kg intravenously three times a week (BTZ group; n = 25); the last group was treated with BTZ 0.2 mg/kg intravenously three times a week plus CR4056 6 mg/kg/day orally (BTZ-CR4056 group; n = 8). After 6 weeks of treatment, at the beginning of phase 2, all groups maintained the initial treatment except the rats of the BTZ group that were re-randomized on the basis of the results obtained from the dynamic test into three groups: the first group continued on BTZ 0.2 mg/kg intravenously three times a week (BTZ group; n = 8); the second was treated with BTZ 0.2 mg/kg intravenously three times a week plus CR4056 6 mg/kg/day orally during the whole 4-week period of phase 2 (BTZ + CR4056 4ws group; n = 8); while the last group of rats was treated with BTZ 0.2 mg/kg intravenously three times a week plus CR4056 6 mg/kg/day orally only during the last 2 weeks of phase 2 (BTZ + CR4056 2ws group; n = 9). (See for flow charts of Experiments 1, 2, and 3.)
Figure 1 Flow charts of in vivo experiments: Experiments 1, 2, and 3.
General toxicity
General toxicity was monitored by daily clinical observation of the rats and the body weight assessments. Body weight was recorded twice weekly and the drug dose was adjusted to the nearest recorded value. Body weight was also recorded on the day of sacrifice.
Hematological and blood chemistry determinations
At the end of the treatment phase, whole blood was obtained from all rats through abdominal aorta puncture, collected into a heparinized tube for complete blood cell determination with an ABX Pentra 60 C+ device (Horiba ABX Diagnostic, Montpellier, France). Serum was obtained by centrifugation of clotted blood at 2.500 g for 15 minutes at 4°C and used for blood urea nitrogen, creatinine, aspartate transaminase, and alanine amino transferase determination (Mira® Plus; Horiba ABX Diagnostic).Citation28
Neurotoxicity
Nerve conduction velocity (NCV) was evaluated after 8 weeks of treatment with BTZ and at the end of Experiments 1 and 2. Neurophysiologic assessment was determined in the tail of each rat as previously described in several experimental paradigms.Citation25,Citation29,Citation30 Briefly, the NCV was assessed by placing recording ring electrodes distally in the tail, while the stimulating ring electrodes were placed 5 and 10 cm proximally with respect to the recording point. The latencies of the potentials recorded at the two sites after nerve stimulation was determined (peak to peak) and NCV was calculated accordingly. All the neurophysiologic determinations were performed under standard conditions in a temperature-controlled room and with vital parameter monitoring of the rats.
Behavioral tests
Two behavioral tests (plantar and dynamic) were used to determine the alterations in pain perception related to chronic treatment with BTZ.
A Plantar Test (Hargreaves’s Apparatus) (No 37370; Ugo Basile Biological Instruments, Comerio, Italy) was used to evaluate the response to a noxious thermal stimulus. The plantar test was performed only during Experiment 1 at baseline and subsequently after 4 and 8 weeks of treatment (phase 1). During phase 2, the plantar test was repeated on day 7, 3 hours or 30 minutes after CR4056 or Bupre administration, respectively. Rats from all groups were accustomed to the test 3 days before performing the baseline evaluation, and during the plantar test rats were placed into a Plexiglas® chamber (28 × 40 × 35 cm, glass floor) for a 15-minute acclimation period followed by testing. A movable 40 W infrared radiant heat source was placed directly under the plantar surface of the hind paw and the time taken for hind paw withdrawal was monitored automatically (withdrawal latency). The mean value of four repeated measures represented the nociceptive threshold. In all experiments, animal safety was guaranteed by a cutoff period of 30 seconds.
The mechanical nociceptive threshold was assessed using a Dynamic Plantar Aesthesiometer (No 37450; Ugo Basile Biological Instruments), which generated a linearly increasing mechanical force.Citation31 In Experiment 1 the dynamic test was performed at baseline and subsequently after 4 and 8 weeks of treatment (phase 1). During phase 2, when the effect of the test compound was assessed, the dynamic test was repeated on days 1, 3, 7, and 14. In Experiment 2, during phase 1, the dynamic test was performed at baseline and subsequently after 8 weeks. During phase 2, the dynamic test was repeated on days 1, 4, 14, and 21 and at the end of the washout period. In both experiments, a single investigator performed the dynamic test 3 hours, 1 hour, or 30 minutes after CR4056, Gaba, or Bupre administrations, respectively. Before performing the test at baseline, rats were adapted to the full technique for 30 minutes a day on 2 consecutive days and testing was conducted on the third day. On each day of testing, rats were placed in a Plexiglas chamber (28 × 40 × 35 cm, wire mesh floor) for a 15-minute acclimatization period followed by testing. After the acclimatization period, a servo-controlled mechanical stimulus, represented by a pointed metallic filament of 0.5 mm diameter, was positioned under the plantar surface of the hind paw, exerting a progressively increasing punctate pressure, reaching up to 50 g within 20 seconds. The pressure bringing a clear spontaneous hind paw withdrawal response was recorded automatically and taken as representing the mechanical nociceptive threshold index. The mechanical threshold was always assessed alternatively on each paw every 2 minutes on three occasions to obtain a mean value. The results evidenced the maximal pressure (expressed in grams) tolerated by the rats. If paw movement subsequent to stimulus initiation appeared to be associated with grooming or locomotion, that measure was repeated after a delay of 1 minute. In order to ensure animal safety, an upper limit cutoff of 30 seconds was set, after which time the mechanical stimulus was automatically ended.
Histopathological examination
At the end of the study period the rats were sacrificed by CO2 inhalation. The left sciatic nerves were removed, fixed by immersion in 3% glutaraldehyde, postfixed in osmium tetroxide, embedded in epoxy resin, and used for light microscope observations. Semithin sections (1.5 μm thick) were prepared from at least two tissue blocks for each animal. The sections were stained with toluidine blue and examined with a Coolscope® light microscope (Nikon Instruments SpA, Calenzano, Italy).
Another group of rats belonging to each experimental group were sacrificed under general anesthesia. The L4–L5 dorsal root ganglia (DRG) were obtained from each animal, processed according to previously reported protocols, and observed at light microscopy.Citation29,Citation30,Citation32,Citation33 Semithin sections (1.5 μm thick) were prepared from at least two tissue blocks for each animal. The sections were stained with toluidine blue and examined with a Coolscope light microscope.
In vitro studies
Cytotoxicity: chemicals
RPMI (Roswell Park Memorial Institute) 1640 medium, penicillin (100 U/mL), streptomycin (100 μg/mL), and HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) were from EuroClone SpA (Pero, Italy). Fetal bovine serum (FBS) was from Hyclone Laboratories, Inc (Logan, UT); sodium bicarbonate and 2-mercaptoethanol were from GIBCO (Carlsbad, CA); and sodium pyruvate was from Lonza (Verviers, Belgium). All other chemicals were from Sigma-Aldrich (St Louis, MO).
Cytotoxicity: cell lines
All cell lines used in this study were a kind gift from Dr Maurizio D’Incalci (Mario Negri Institute for Pharmacological Research, Milan, Italy).
The human non-small cell lung cancer cell lines NCI-H460 (H460) and A549 (adenocarcinoma) were maintained as monolayer cultures in RPMI medium (containing 2 mM L-glutamine) supplemented with 10% FBS, penicillin, and streptomycin. Cells were grown in 75 cm2 culture flasks (Corning Incorporated, Corning, NY), at 37°C in 5% CO2 and 95% air, and harvested with trypsin-ethylenediaminetetraacetic acid when they were in exponential growth.
The human RPMI 8226 MM cells were maintained in floating culture in RPMI medium (containing 2 mM stable L-glutamine) supplemented with 10% FBS, penicillin, and streptomycin. The human H929 MM cell line was maintained in floating culture in RPMI medium (containing 2 mM stable L-glutamine) supplemented with 10% FBS, penicillin, streptomycin, 1.5 g/L sodium bicarbonate, 10 mM HEPES, 1 mM sodium pyruvate, and 0.05 mM 2-mercaptoethanol.
Cytotoxicity assay
The MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay was used to study the cell growth inhibitory effect of BTZ. Cells were plated in 96-well plates (Corning Incorporated): A549 and H460 cells at 3000 cells/well, RPMI 8226 and H929 cells at 20000 cells/well. After 24 hours cells were treated with BTZ (0.05–250 nM) for 72 hours. At the end of the incubation, cells were incubated with MTT (final concentration, 0.5 mg/mL); formazan crystals were dissolved in dimethyl sulfoxide for A549 and H460 cells or in 0.04 N hydrochloric acid in isopropanol for RPMI 8226 and H929 cells. The optical density was then measured at 540 nm. Growth inhibition was expressed as the percentage of CTRL (vehicle-treated cells) absorbance, corrected for absorbance before drug addition. The 50% inhibitory concentration (IC50) of cell growth was calculated by nonlinear least squares curve fitting, using GraphPad Prism software (v 3.0; GraphPad Software Inc, La Jolla, CA).
Drug combination studies
To rule out any interference of CR4056 with BTZ-induced cytotoxicity, non-small cell lung cancer and MM cell lines were simultaneously treated for 72 hours with BTZ and CR4056. Cells were exposed to the IC50 of BTZ, estimated in the cytotoxicity study described earlier, with or without three concentrations of CR4056 (3, 10, and 30 μM). The incubations with CR4056 alone (3, 10, and 30 μM) and with the highest dose of vehicle (DMSO) were also performed. Growth inhibition was assessed by MTT assay, as described earlier. In these experiments, the CR4056 was tested in vitro in a range of concentrations spanning from pharmacological to toxicological levels. In particular, the lowest concentration (3 μM) was approximately fivefold the affinity of CR4056 for I2 sites (IC50 = 0.6 μM)Citation22 and the highest concentration (30 μM) was in the range of plasma levels found in rats treated with 200 mg/kg of CR4056 during a 28 day repeated dose toxicology study (maximum plasma concentration [Cmax] = 32 μM) (unpublished data on file).
Statistical evaluation
All data are expressed as mean plus or minus standard deviation. In both in vivo and in vitro studies, the differences between all experimental groups were statistically evaluated using the analysis of variance and the Tukey-Kramer post hoc test (significance level set at P < 0.05), using GraphPad Prism software.
Results
In vivo studies on BTZ-induced neuropathy
General toxicity
The administration of BTZ was fairly well tolerated. Serious adverse events, leading to death, were limited to about 5% of total BTZ-treated rats. In particular, during phase 1 of Experiments 1, 2, and 3 some rats died after the first administration – four, two, and two, respectively – and reserve animals were used to immediately replace these rats. During phase 2 of Experiments 1 and 3, a further number of rats died during the treatment – three and one, respectively – and these rats were not replaced; all these rats were allocated to groups treated only with BTZ. The changes in number within groups at the end of phase 2 are recorded in .
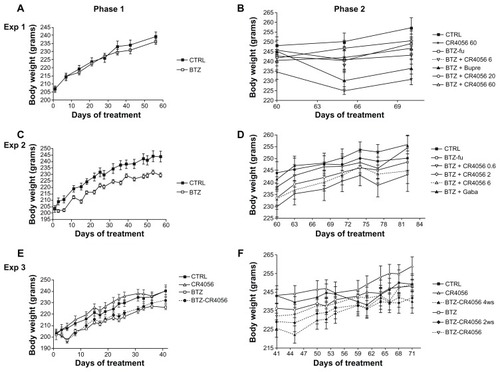
The oral administration of all doses of CR4056 was well tolerated in all experiments and no mortality was observed. shows the weight changes during the different phases of Experiments 1, 2, and 3. In Experiment 1 no significant difference was observed among groups at baseline or at the end of phase 1 (). In contrast, in Experiments 2 and 3, BTZ marginally but significantly affected body weight growth (). When CR4056 was coadministered with BTZ, as in Experiment 3, it neither worsened nor improved the toxicity induced by BTZ. No significant changes were observed in each group between the values measured during phase 2 ().
Figure 2 Body weight changes along the study period: Experiment (Exp) 1, phase (A) 1 and (B) 2; in Exp 1, bortezomib (BTZ)-treated animals do not show any significant difference in weight gain with respect to the control (CTRL) at the end of the 8-week treatment. Exp 2, phase (C) 1 and (D) 2; Exp 3, phase (E) 1 and (F) 2; in Exps 2 and 3, BTZ significantly affected body weight growth (C and E) (P < 0.01); CR4056 when coadministered with BTZ neither worsened nor improved the toxicity induced by BTZ (E). No significant changes were observed in each group between the values measured during phase 2 (B, D, and F).
Abbreviations: BTZ-fu, bortezomib-fake-manipulated but untreated; Bupre, buprenorphine; CR4056 0.6, CR4056 at 0.6 mg/kg/day; CR4056 2, CR4056 at 2 mg/kg/day; CR4056 20, CR4056 at 20 mg/kg/day; CR4056 6, CR4056 at 6 mg/kg/day; CR4056 60, CR4056 at 60 mg/kg/day; Gaba, gabapentin; ws, weeks.
The hematological and blood chemistry analyses were performed at the end of phase 1 and 2 of all experiments. No remarkable effect on the examined hematological and blood chemistry parameters resulted from BTZ administration, or from any dose of CR4056 (even when coadministered with BTZ in Experiment 3), Bupre, or Gaba (data not shown).
Neurophysiologic assessment
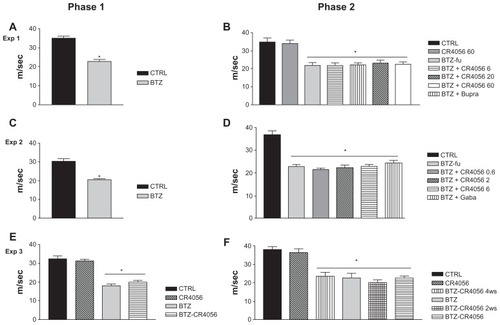
The results of NCV determinations performed during Experiments 1, 2, and 3 are reported in . In all the experiments the neurophysiologic determinations evidenced a statistically significant reduction in NCV in BTZ-treated rats (P < 0.001, BTZ versus CTRL rats) at the end of the 8-week treatment period during phase 1 (). At the end of phase 2, no effect of CR4056 at any dose was observed in the subsequent NCV determinations (), even when co-treatment with CR4056 (6 mg/kg/day) and BTZ was started from the beginning of phase 1 (). Similarly, Bupre or Gaba administration had no effect on the NCV impairment induced by BTZ administration. The same results were confirmed at the end of the follow-up period (data not shown).
Figure 3 Results of nerve conduction velocity (NCV) study performed on the tail of control (CTRL) and bortezomib (BTZ)-treated rats: Experiment (Exp) 1, phase (A) 1 and (B) 2; Exp 2, phase (C) 1 and (D) 2; Exp 3, phase (E) 1 and (F) 2. BTZ induced a significant reduction in NCV in all BTZ-treated rats after 8 weeks of treatment and at the end of Exps 1, 2, and 3.a The analgesic compounds tested did not significantly counteract the effect of BTZ on NCV.
Abbreviations: BTZ-fu, bortezomib-fake-manipulated but untreated; Bupre, buprenorphine; CR4056 0.6, CR4056 at 0.6 mg/kg/day; CR4056 2, CR4056 at 2 mg/kg/day; CR4056 20, CR4056 at 20 mg/kg/day; CR4056 6, CR4056 at 6 mg/kg/day; CR4056 60, CR4056 at 60 mg/kg/day; Gaba, gabapentin; ws, weeks.
Thermal nociception (plantar test)
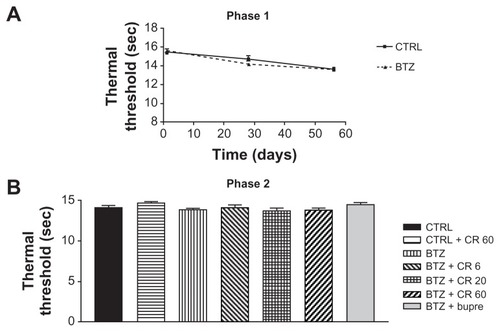
Thermal algesia was only evaluated in Experiment 1, during both phase 1 and phase 2. No difference was observed between BTZ-treated and untreated groups at the end of phase 1. The administration of Bupre or CR4056 in phase 2 had no significant effect at any dose ().
Figure 4 Plantar test phase 1 (A) and 2 (B) in Experiment 1: bortezomib (BTZ) and CR4056 (CR) did not induce any significant modification in thermal threshold with respect to the control (CTRL) rats at any experimental time point evaluated in this study.
Abbreviations: Bupre, buprenorphine; CR 20, CR4056 at 20 mg/kg/day; CR 6, CR4056 at 6 mg/kg/day; CR 60, CR4056 at 60 mg/kg/day; sec, second.
Mechanical allodynia (dynamic test)
In contrast with the thermal algesia findings, rats treated with BTZ developed a significant mechanical allodynia at the end of phase 1 in all the experiments performed.
All data obtained in the dynamic test at baseline (ie, before starting BTZ treatment, rats aged 8–9 weeks) and at the end of phase 1 (8 weeks for Experiments 1 and 2; 6 weeks for Experiment 3) are summarized in .
Table 1 Dynamic test data (mechanical nociceptive threshold in grams) at baseline (ie, before starting bortezomib [BTZ] treatment) and at the end of phase 1 (8–6 weeks)Table Footnotea
In Experiment 3 a group of rats was co-treated with CR4056 (6 mg/kg/day) and BTZ from the beginning of phase 1, these rats were fully protected against BTZ-induced allodynia (), in spite of the lack of efficacy in counteracting the reduction in NCV induced by BTZ (see “Neurophysiologic assessment” section).
In Experiment 1, during the whole phase 2 period of observation (2 weeks – ie, BTZ free and test compound treated), BTZ-fu rats showed dynamic test values significantly lower than those of CTRL rats (), thus demonstrating a stable level of allodynia. In this experiment, the oral administration of CR4056 at 20 or 60 mg/kg had a significant effect on days 1 and 3 (P < 0.001, as compared with BTZ-fu group), but not on days 7 and 14. In contrast, CR4056 at 6 mg/kg was effective along the entire phase 2 period of the study (P < 0.05 on days 1, 7, and 14; P < 0.001 on day 3, as compared with BTZ-fu group) (). The administration of Bupre during phase 2 had a significant effect only on day 1 (P < 0.05, as compared with BTZ-fu group), since this effect was no longer evident at the subsequent examinations.
Figure 5 Dynamic test in phase 2 of (A) Experiment (Exp) 1a; (B) Exp 2b; and (C) Exp 3c. The persistent and significant antiallodynic effect of oral CR4056 6 mg/kg was evident in all studies, in comparison with buprenorphine (Bupre), gabapentin (Gaba), and the other doses of CR4056. Before CR4056 administration, in Exps 1, 2, and 3, bortezomib (BTZ)-treated rats showed a significant difference compared with control (CTRL) (P < 0.05).
Abbreviations: admin, administration; BTZ-fu, bortezomib-fake-manipulated but untreated; CR4056 0.6, CR4056 at 0.6 mg/kg/day; CR4056 2, CR4056 at 2 mg/kg/day; CR4056 20, CR4056 at 20 mg/kg/day; CR4056 6, CR4056 at 6 mg/kg/day; CR4056 60, CR4056 at 60 mg/kg/day; Gaba, gabapentin; IV, intravenously; OS, orally; tiw, three times a week; ws, weeks.
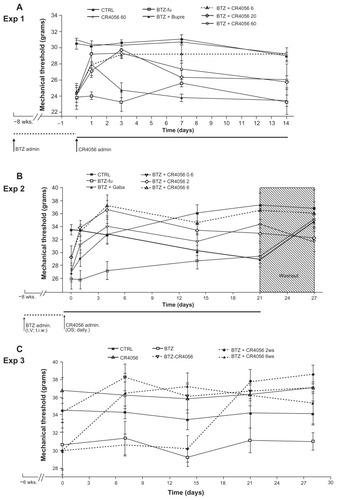
In Experiment 2, which had the longer follow-up period of observation (4 weeks), allodynia in BTZ-fu rats consistently lasted until day 21 (P < 0.05, as compared with CTRL group), but it was no longer observed at day 28 after BTZ withdrawal (). In this experiment, the oral administration of CR4056 at 0.6, 2, and 6 mg/kg showed a dose-related and statistically significant antiallodynic effect (P < 0.01 for BTZ + CR4056 2 and 6 groups, as compared with BTZ-fu group; P < 0.05 for BTZ + CR4056 0.6 group, as compared with BTZ-fu group) (). The analgesic effect (approximately 100% of the maximum possible effect) at the dose of 6 mg/kg was persistent and statistically significant also at the end of the second and third week of treatment, confirming the lack of tolerance at this dose. Moreover, the suspension of CR4056 treatment after day 21 did not induce any sign of withdrawal syndrome, since no hyperalgesic rebound was observed. The administration of Gaba (100 mg/kg subcutaneously) during phase 2 induced a mild acute effect on the first day that attained statistical significance only after the fourth administration; however, this effect was no longer evident at the subsequent examinations on days 14 and 21.
Experiment 3 evidenced that the analgesic effect of a daily oral administration of 6 mg/kg CR4056 was observed both in preventive and therapeutic schedules of treatment without developing tolerance. Moreover, the starting time of CR4056 administration in the rats treated only with BTZ for 6 or 8 weeks did not change the efficacy of the CR4056 analgesic effect: the reversion of BTZ-induced allodynia was complete and lasted for all phase 2.
Sciatic nerve and DRG specimens examination
Sciatic nerves and DRG were collected in Experiments 1 and 2 at the end of the study. The light microscope morphological analysis of the sciatic nerve showed that the administration of BTZ induced mild axonopathy, while degeneration of the myelin sheath was very occasionally observed. Axonopathy was predominant in small myelinated and unmyelinated fibers, although occasionally also large myelinated fibers were affected. In the DRG, neurons were spared by the neurotoxic effect of BTZ. By contrast, the administration of BTZ induced mild morphological alteration in a proportion of DRG satellite cells. These changes were overall rather mild and they were represented by the presence of vacuoles in the cytoplasm. These pathological results were consistent with those already extensively described in the same model.Citation27 The qualitative examination evidenced no differences attributable to the administration of CR4056 in treated groups. Also the administration with Bupre or Gaba, in Experiments 1 and 2, respectively, did not modify the pathological alterations induced by BTZ (data not shown).
In vitro studies to evaluate if CR4056 interferes with BTZ-induced cytotoxicity
Cytotoxicity of bortezomib
BTZ induced a concentration-dependent inhibition of cell growth in all the tested cell lines. In particular, H929 myeloma cells were the most sensitive cell line after 72-hour exposure to BTZ, (IC50 = 1.0 ± 0.25 nM). The IC50 BTZ values for A549, RPMI 8226, and H460 cell lines were 2.4 ± 0.3 nM, 4.5 ± 0.8 nM, and 15 ± 0.6 nM, respectively.
Combination study
H929, A549, RPMI 8226, and H460 cell lines were exposed for 72 hours to BTZ concentrations able to induce IC50. The co-treatment with CR4056 at all the concentrations used (range 3–30 μM) did not induce any significant difference in cell survival compared with BTZ-treated cells, either in H929 or in RPMI 8226 myeloma cells ().
Figure 6 Cytotoxicity effect of bortezomib (BTZ) and CR4056 in (A) A549, (B) H460, (C) RPMI 8226, and (D) H929 cell lines.
Abbreviation: DMSO, dimethyl sulfoxide.
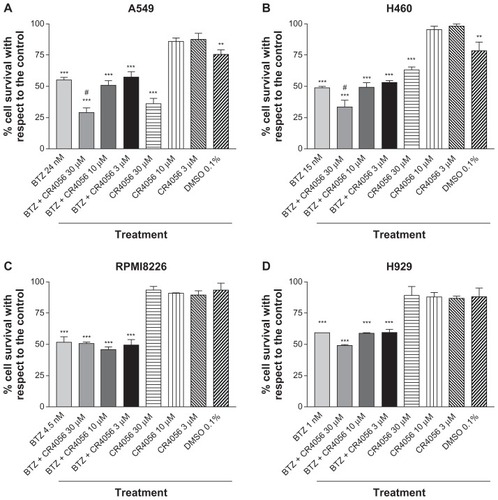
Conversely, the treatment of A549 and H460 cells with BTZ and CR4056 at the highest concentration used in this study (30 μM) induced a significant decrease in cell survival (P < 0.01, as compared with BTZ-treated cells). This was probably due, at least in part, to the high concentration of DMSO (0.1%) used to dissolve the highest CR4056 concentration. In fact, 0.1% DMSO in the absence of either BTZ or CR4056 induced a significant decrease in cell survival in both A549 and H460 cells (P < 0.01, as compared with untreated cells). However, on the basis of the current data, a direct effect of 30 μM CR4056 on A549 and H460 cells cannot be ruled out. The 0.1% DMSO-induced toxicity was not observed in MM cell lines (H929 and RPMI 8226).
Discussion
BTZ therapy is currently proposed as the standard of care for the treatment of MM.Citation34,Citation35 Unfortunately, the clinical administration of this highly effective anticancer agent is almost invariably associated with the induction of a painful peripheral neuropathy as a major disabling adverse effect.Citation13 This NP induced by BTZ is difficult to manage in the clinical practice with standard analgesics, and the use of currently available drugs able to block abnormal pain sensation is based largely on empirical evidence.Citation36,Citation37 Although the physiological mechanisms of their antinociceptive effect are still not fully understood,Citation38–Citation40 the actual clinical guidelines suggest anticonvulsants such as Gaba, together with some antidepressants, as first-line drugs for NP treatment.Citation41,Citation42
In the last few years a model of BTZ-induced peripheral neuropathy was successfully established in the rat.Citation25 This model was selected because of the significant mechanical allodynia observed after BTZ treatmentCitation27 that resembles the human neuropathy,Citation43,Citation44 even when no thermal hyperalgesia can be evidenced. Furthermore, a BTZ treatment schedule in the rat induced degeneration of myelinated fibers, with damage of satellite cells and DRG neurons.Citation25,Citation45 The present study demonstrates for the first time the analgesic efficacy of CR4056 in counteracting the mechanical allodynia induced by BTZ in this pain model. Moreover, this study shows that the in vitro co-treatment with CR4056 did not hinder the cytotoxic effect of BTZ in MM cell lines, suggesting no interference with the in vivo antitumor activity of BTZ.
CR4056 is a new synthetic compound belonging to the emerging class of imidazoline I2 ligandsCitation18 showing an outstanding oral efficacy in several murine models of inflammatory and NP.Citation22 Previous results suggest that compounds of this class, being able to cross the blood–brain barrier efficiently, may both attenuate the development of opioid tolerance and dependence and control the central sensitization from higher centers (eg, locus coeruleus) to spinal nociceptors.Citation46 Nevertheless, the exact physiological mode of action involved in the control of pain by I2 selective ligands is far from being fully elucidated. In fact, on the basis of current literature, at least a dual mechanism could be assumed. First, recent evidenceCitation24 has shown that agmatine, the putative endogenous ligand of I2 receptors, can mimic some of the effects of opioid receptor ligands by inducing beta-endorphin release from the adrenal medulla. CR4056 is devoid of affinity towards opiate receptors in vitro.Citation22 Moreover, the authors have never observed a direct activation of the opiate system in vivo: no myosis, constipation, or opiate-like behaviors (eg, Straub tail), even at toxicological doses. Furthermore, the authors never recorded a withdrawal syndrome, either in pharmacological tests (the present study’s Experiment 2) or after stopping drug administration in the recovery animals during the repeated dose toxicological studies. However, the implication of endogenous opiate activation could not be excluded from the mode of action of CR4056 – that is, as discussed earlier, a common feature of I2 ligands. For instance, as discussed by Ferrari et alCitation22 naloxone is able to abrogate the hypoalgesic component of the activity of CR4056 (but not the frank analgesic effect), even if the strong synergy demonstrated by CR4056 with morphineCitation22 seems to completely preclude the two compounds acting on the same target for their analgesic activity.
The second putative mode of action assumed for I2 ligands in the control of pain is based on experimental evidence describing the allosteric modulation by both agmatine and other I2 selective ligands on MAO activity.Citation47 This class of enzymes is largely responsible for monoamine catabolism, and is heavily expressed on small-diameter sensory neuronsCitation48 and some nociceptive primary afferent neurons.Citation21 In particular, the selective inhibition of MAO-A increases the synaptic levels of noradrenaline and serotonin by inhibiting their degradation. The role of these catecholamines in controlling pain is well documented by both preclinical and clinical data.Citation49,Citation50 CR4056 selectively inhibits the activity of the human recombinant MAO-A in a concentration-dependent manner,Citation22 as well the murine isoform in the submicromolar range. Accordingly, the oral administration of CR4056 induced a significant increase of noradrenaline tissue levels both in the cerebral cortex and in the lumbar spinal cord.Citation22 Since the increased synaptic availability of noradrenaline and/or serotonin might be considered as the precondition for a drug to exert an antidepressant-like activity, the authors tested CR4056 in murine behavioral models of depression such as the Porsolt forced swimming test and the tail suspension test () and in the same range of doses active in NP models. Indeed, CR4056 confirmed its potential to modulate the catecholaminergic transmission by dose-dependently decreasing the immobility time of mice in either of the experimental paradigms.
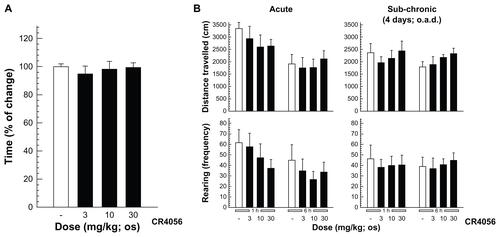
Recently, the hypothermic effects of imidazoline I2 receptor ligands including CR4056 in rats have been reported. Citation51 Moreover, the study by Thorn et alCitation51 has shown that CR4056, when acutely administered intraperitoneally in the range of 10–56 mg/kg, apparently and significantly decreases locomotor activity in rats. These effects could potentially influence the endpoint behavior of the present study in nociceptive tests. However, as demonstrated by Ferrari et alCitation22 and re-analyzed in the present paper (), no evidence of significant inhibition of locomotor activity was found after acute and/or subchronic oral administrations of CR4056 in the range of 3–30 mg/kg. Moreover, during regulatory central nervous system safety pharmacology studies, CR4056 was tested orally at doses of 50, 160, and 500 mg/kg, and both the Irwin standardized observation battery and the body temperature were monitored. No neurobehavioral changes were considered related to CR4056 administration, at any dose level. However, a statistically significant decrease in body temperature was noted at 500 mg/kg between 2 and 6 hours post dose. The calculated “no observed effect level” for CR4056 was considered to be 160 mg/kg in this study. From a pharmacokinetic point of view, Rottapharm internal data demonstrated that after an intraperitoneal injection of 30 mg/kg of CR4056, a plasma Cmax value of approximately 30 μg/mL is rapidly reached and is maintained for several hours. On the other hand, during pharmacodynamic/pharmacokinetic and toxicokinetic studies the oral administration of doses of CR4056 encompassing the range of effective doses against BTZ- induced neuropathy in this paper (2–60 mg/kg) produces a Cmax ranging from 0.7 to 6 μg/mL (approximately 1 μg/mL with the optimal 6 mg/kg dose). Moreover, the “no observed adverse effect level” in the Irwin test (160 mg/kg) was associated with a Cmax of 10 μg/mL, while the body temperature-lowering dose of 500 mg/kg had a Cmax of 20 μg/mL in the range of what was presumably obtained by Thorn et alCitation51 upon intraperitoneal administration. Therefore, the effect described by Thorn et alCitation51 is probably a suprapharmacological effect, linked to I2 occupancy but without any interference with the effectiveness of CR4056 against BTZ-induced hyperalgesia.
Since the mechanisms underlying BTZ-induced painful neuropathy are largely unknown, it is impossible to determine the mode of action of CR4056 in modulating this side effect of the drug. However, on the basis of previous observations indicating satellite cell changes in the DRG of BTZ-treated animalsCitation25,Citation27 and of the well-established involvement of this cell population in NP,Citation52 the authors carefully searched for any morphological effect induced in DRG glial cells in CR4056 co-treated rats, without evidence of any difference, compared with animals treated with BTZ alone. It is also important to underline that CR4056, when administered at pharmacological doses, did not impair the normal locomotor activity, as already described by Ferrari et alCitation22 (). Moreover, as required by regulatory guidelines, the authors had performed a good laboratory practice–compliant regulatory behavioral study (Irwin test) at suprapharmacological doses (50, 160, and 500 mg/kg) in the safety pharmacology package enabling the first administration to humans. Under the experimental conditions adopted, no neurobehavioral changes that were considered related to CR4056 administration were noted at any dose level. Of particular value, in this context, is the lack of signs of impaired awareness (including catalepsy) or locomotor activity (including spontaneous locomotion) and of disturbed muscle tone (including hypotonic gait), even after the highest dose tested (500 mg/kg – ie, approx 100-fold the optimal dose in BTZ-induced neuropathy).
In this study the BTZ-treated rats were found to show a significant reduction in the NCV and the development of a relevant and persistent mechanical allodynia. The administration of all the tested doses of CR4056 (including the lowest dose of 0.6 mg/kg) exerted a significant analgesic effect, but only the oral daily dose of 6 mg/kg fully abrogated BTZ-induced allodynia along the entire period of administration. It is worth noting that CR4056 efficacy on BTZ-induced allodynia was invariably observed, irrespective of the kind of protocol (ie, CR4056 coadministered with BTZ from the beginning of the experiment or only when allodynia was already established, as in Experiment 3, or administered alone after BTZ cessation, as in Experiments 1 and 2). Moreover, in this model, the effect of CR4056 seems purely analgesic, since the product was unable to counteract the impaired conduction velocity induced by BTZ, even when CR4056 was coadministered with BTZ from the very beginning of the experiment.
Bupre and Gaba, administered at rat doses equivalent to the human therapeutic doses did not show a similar constant analgesic effect, since both compounds exerted their effect only for a few days after administration. Although it is possible that the selected doses of Gaba and Bupre were not optimal to evidence a significant reduction of mechanical allodynia in this rat model of pain,Citation53 they showed in other models of NP to be within the range of analgesic activity of these drugs.Citation29,Citation54
In view of the very promising analgesic activity of CR4056 seen in the present study against BTZ-induced NP and of a possible future clinical development of this product in oncological patients, the authors were prompted to test if CR4056 could somehow impair the antitumor efficacy of BTZ. Therefore, several in vitro tests were performed at suprapharmacological concentrations of CR4056 to rule out this possibility. In fact, the in vitro tests on BTZ-induced cytotoxicity in two MM cell lines (RPMI 8226 and H929) and two solid tumor cell lines sensitive to BTZ (H460 and A549 lung cancer cell lines) showed that CR4056, over the range of concentrations of 3–30 μM, was unable to hinder BTZ cytotoxicity on these tumor cell lines, which could indicate that this substance does not directly interfere with BTZ antitumor activity.
Conclusion
The results suggest a role for CR4056 in the modulation of allodynic pain after BTZ treatment, and when taken together, CR4056 and BTZ demonstrate a long-lasting effect of CR4056 in this NP model. Although further investigations are needed to evaluate the effect of CR4056 in preventing (preventive schedule) or reversing (therapeutic schedule) the NP induced by a subchronic treatment with BTZ, the authors believe that the administration of CR4056 may have the potential to be an innovative analgesic therapy for BTZ-treated patients. Moreover, in light of the data reported here, a possible role for CR4056 in the treatment of other chemotherapy-induced peripheral neuropathies deserves to be evaluated.
Acknowledgments/disclosure
The authors wish to thank Dr Elisa Ballarini, Dr Virginia Rodriguez-Menendez, and Mario Bossi for their assistance with the histological evaluation.
This work was supported in part by the Italian Ministry of Health grant “Progetto di Ricerca Oncologica 2006 – Assessment and rehabilitation in patients with chemotherapy-induced peripheral neuropathy” in collaboration with the “Fondazione Ospedale San Camillo – Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS).”
ML, OL, FF, and GFC are full-time employees of Rottapharm SpA (Monza, Italy). Rottapharm SpA is developing CR4056 as an analgesic drug. The product is not marketed yet; Rottapharm SpA has filed the following patent applications on CR4056: WO 2008/014822, WO 2009/152868, and WO 2010/140139. However, the Rottapharm group as a corporate entity had no role in the conduct of the study; in the collection, analysis, and interpretation of data; in the writing of the report; or in the decision to submit the paper for publication.
Supplementary figures
Figure S1. Chemical structure of CR4056.
Figure S2. (A) Porsolt forced swimming test: CR4056 (3, 10, and 30 mg/kg) was administered orally (per os) to male CD1 mice 1 hour before the test and both the immobility time and the latency to immobility were evaluated. (B) Tail suspension test: CR4056 was administered per os to male CD1 mice 30 minutes before the test; two independent experiments were performed to cover a broad range of doses (from 0.1 to 30 mg/kg).
Abbreviations: ns, not significant; sec, second.
Notes: *P < 0.05; ** P < 0.01.
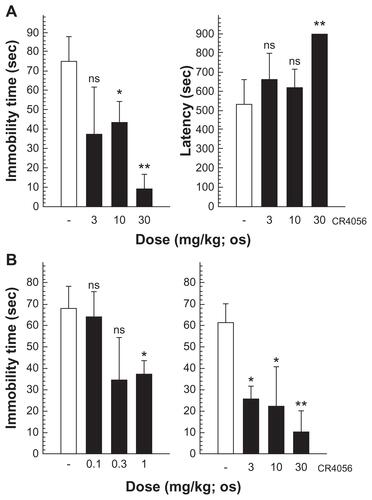
Figure S3. (A) Rota-rod assay: rats were allowed a 30-minute acclimatization period in the testing room and then placed on a 9 cm diameter rod, which increased the speed from 0 to 20 rpm over a 60-second period. The time required for the rat to fall from the rod was recorded, with a maximum score of 60 seconds.a (B) Open field test: CR4056 effects in the open field assay data (evaluated 1 and 6 hours after oral administration) are expressed as the distance traveled and as the rearing activity following an acute (left) and a subchronic treatment period (right).b
Notes: aMean plus or minus standard error of the mean (n = 5 rats/group); bmean plus or minus standard error of the mean (n = 6 rats/group).
Abbreviations: h, hour; os, orally; o.a.d., once a day.
References
- KannarkatGLasherEESchiffDNeurologic complications of chemotherapy agentsCurr Opin Neurol200720671972517992096
- KuroiKShimozumaKNeurotoxicity of taxanes: symptoms and quality of life assessmentBreast Cancer2004111929914718800
- WindebankAJGrisoldWChemotherapy-induced neuropathyJ Peripher Nerv Syst2008131274618346229
- CataJPWengHRLeeBNReubenJMDoughertyPMClinical and experimental finding in humans and animals with chemotherapy-induced peripheral neuropathyMinerva Anestesiol200672315116916493391
- BennettGJNeuropathic painWallPDMelzackRTextbook of Pain3rd edLondonChurchill Livingston1994201220
- CampbellJNMeyerRAMechanisms of neuropathic painNeuron2006521779217015228
- AdamsJPalombellaVJSausvilleEAProteasome inhibitors: a novel class of potent and effective antitumor agentsCancer Res199959112615262210363983
- CurranMPMcKeageKBortezomib: a review of its use in patients with multiple myelomaDrugs200969785988819441872
- TobinaiKProteasome inhibitor, bortezomib, for myeloma and lymphomaInt J Clin Oncol200712531832617929113
- KennedyBGargoumFBystrickyBCurranDRO’ConnorTMNovel agents in the management of lung cancerCurr Med Chem201017354291432520939812
- HideshimaTChauhanDRichardsonPNF-kappa B as a therapeutic target in multiple myelomaJ Biol Chem200227719166391664711872748
- LenzHJClinical update: proteasome inhibitors in solid tumorsCancer Treat Rev200329Suppl 1S41S48
- CavalettiGNobile-OrazioEBortezomib-induced peripheral neurotoxicity: still far from a painless gainHaematologica200792101308131018024368
- PariniAMoudanosCGPizzinatNLanierSMThe elusive family of imidazoline binding sitesTrends Pharmacol Sci199617113168789353
- FinnDPMartíOHarbuzMSBehavioral, neuroendocrine and neurochemical effects of the imidazoline I2 receptor selective ligand BU224 in naive rats and rats exposed to the stress of the forced swim testPsychopharmacology (Berl)2003167219520212652345
- HeadGAMayorovDNImidazoline receptors, novel agents and therapeutic potentialCardiovasc Hematol Agents Med Chem200641173216529547
- LioneLANuttDJHudsonALCharacterisation and localisation of [3H]2-(2-benzofuranyl)-2-imidazoline binding in rat brain: a selective ligand for imidazoline I2 receptorsEur J Pharmacol199835311231359721049
- LiJXZhangYImidazoline I2 receptors: target for new analgesics?Eur J Pharmacol20116582–3495621371460
- EglenRMHudsonALKendallDA‘Seeing through a glass darkly’: casting light on imidazoline ‘I’ sitesTrends Pharmacol Sci19981993813909786027
- MattiaCColuzziFIndantadol, a novel NMDA antagonist and nonselective MAO inhibitor for the potential treatment of neuropathic painIDrugs200710963664417786847
- DinaOAKhasarSGAlessandri-HaberNNeurotoxic catecholamine metabolite in nociceptors contributes to painful peripheral neuropathyEur J Neurosci200928611801190 partial retraction in Eur J Neurosci2009;30(11):2235.18783367
- FerrariFFiorentinoSMennuniLAnalgesic efficacy of CR4056, a novel imidazoline-2 receptor ligand, in rat models of inflammatory and neuropathic painJ Pain Res2011411112521647215
- HwangSLLiuIMTzengTFChengJTActivation of imidazoline receptors in adrenal gland to lower plasma glucose in streptozotocin-induced diabetic ratsDiabetologia200548476777515756537
- ChangCHWuHTChengKCLinHJChengJTIncrease of beta-endorphin secretion by agmatine is induced by activation of imidazoline I(2A) receptors in adrenal gland of ratsNeurosci Lett2010468329729919913596
- CavalettiGGilardiniACantaABortezomib-induced peripheral neurotoxicity: a neurophysiological and pathological study in the ratExp Neurol2007204131732517214983
- CantaAChiorazziAMeregalliCContinuous buprenorphine delivery effect in streptozotocine-induced painful diabetic neuropathy in ratsJ Pain200910996196819595641
- MeregalliCCantaACarozziVABortezomib-induced painful neuropathy in rats: a behavioral, neurophysiological and pathological study in ratsEur J Pain201014434335019695912
- CantaAChiorazziACarozziVIn vivo comparative study of the cytotoxicity of a liposomal formulation of cisplatin (Lipoplatin™)Cancer Chemother Pharmacol20116841001100821327681
- CavalettiGCavallettiEMontagutiPOggioniNDe NegriOTrediciGEffect on the peripheral nervous system of the short-term intravenous administration of paclitaxel in the ratNeurotoxicology19971811371459215996
- CavalettiGFabbricaDMinoiaCFrattolaLTrediciGCarboplatin toxic effects on the peripheral nervous system of the ratAnn Oncol1998944434479636837
- MottaEMCalixtoJBRaeGAMechanical hyperalgesia induced by endothelin-1 in rats is mediated via phospholipase C, protein kinase C, and MAP kinasesExp Biol Med (Maywood)200623161141114516741065
- CavalettiGTrediciGMarmiroliPPetruccioliMGBarajonIFabbricaDMorphometric study of the sensory neuron and peripheral nerve changes induced by chronic cisplatin (DDP) administration in ratsActa Neuropathol19928443643711441917
- PersohnECantaASchoepferSMorphological and morphometric analysis of paclitaxel and docetaxel-induced peripheral neuropathy in ratsEur J Cancer200541101460146615913989
- MoreauPPylypenkoHGrosickiSSubcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma: a randomised, phase 3, non-inferiority studyLancet Oncol201112543144021507715
- KortümMEinseleHResults of the first bortezomib-based induction therapy in the treatment of multiple myelomaExpert Opin Pharmacother201112101661166321554151
- ArgyriouAAIconomouGKalofonosHPBortezomib-induced peripheral neuropathy in multiple myeloma: a comprehensive review of the literatureBlood200811251593159918574024
- MohtyBEl-CheikhJYakoub-AghaIMoreauPHarousseauJLMohtyMPeripheral neuropathy and new treatments for multiple myeloma: background and practical recommendationsHaematologica201095231131920139393
- ObataHSaitoSKoizukaSNishikawaKGotoFThe monoamine-mediated antiallodynic effects of intrathecally administered milnacipran, a serotonin noradrenaline reuptake inhibitor, in a rat model of neuropathic painAnesth Analg200510051406141015845695
- SawynokJEsserMJReidARPeripheral antinociceptive actions of desipramine and fluoxetine in an inflammatory and neuropathic pain test in the ratPain199982214915810467920
- McQuayHJTramèrMNyeBACarrollDWiffenPJMooreRAA systematic review of antidepressants in neuropathic painPain1996682–32172279121808
- MoulinDEClarkAJGilronIPharmacological management of chronic neuropathic pain: consensus statement and guidelines from the Canadian Pain SocietyPain Res Manag2007121132117372630
- SaartoTWiffenPJAntidepressants for neuropathic pain: a Cochrane reviewJ Neurol Neurosurg Psychiatry201081121372137320543189
- ArgyriouAABrunaJMarmiroliPCavalettiGChemotherapy-induced peripheral neurotoxicity (CIPN): an updateCrit Rev Oncol Hematol2012821517721908200
- CavalettiGJakubowiakAJPeripheral neuropathy during bortezomib treatment of multiple myeloma: a review of recent studiesLeuk Lymphoma20105171178118720497001
- BrunaJUdinaEAléANeurophysiological, histological and immunohistochemical characterization of bortezomib-induced neuropathy in miceExp Neurol2010223259960820188093
- Ruiz-DurántezETorrecillaMPinedaJUgedoLAttenuation of acute and chronic effects of morphine by the imidazoline receptor ligand 2-(2-benzofuranyl)-2-imidazoline in rat locus coeruleus neuronsBr J Pharmacol2003138349450012569074
- RaddatzRPariniALanierSMImidazoline/guanidinium binding domains on monoamine oxidases. Relationship to subtypes of imidazoline-binding proteins and tissue-specif ic interaction of imidazoline ligands with monoamine oxidase BJ Biol Chem19952704627961279687499273
- VegaJAAmentaFHernandezLCdel ValleMEPresence of catecholamine-related enzymes in a subpopulation of primary sensory neurons in dorsal root ganglia of the ratCell Mol Biol19913755195301682049
- JasminLTienDJanniGOharaPTIs noradrenaline a significant factor in the analgesic effect of antidepressants?Pain20031061–23814581104
- BannisterKBeeLADickensonAHPreclinical and early clinical investigations related to monoaminergic pain modulationNeurotherapeutics20096470371219789074
- ThornDAAnXFZhangYPiginiMLiJXCharacterization of the hypothermic effects of imidazoline I(2) receptor agonists in ratsBr J Pharmacol Epub February 10, 2012
- ScholzJWoolfCJThe neuropathic pain triad: neurons, immune cells and gliaNat Neurosci200710111361136817965656
- KusunoseNKoyanagiSHamamuraKMolecular basis for the dosing time-dependency of anti-allodynic effects of gabapentin in a mouse model of neuropathic painMol Pain201068321108841
- FlattersSJEffect of analgesic standards on persistent postoperative pain evoked by skin/muscle incision and retraction (SMIR)Neurosci Lett20104771434720417687