Abstract
Background
XaraColl®, a collagen-based implant that delivers bupivacaine to the site of surgical trauma, is under development for postoperative analgesia. Because of differing patient attitudes to postoperative pain control and the inability to assess baseline pain, standard clinical methods for evaluating analgesic efficacy are compromised and justify application of novel integrated approaches.
Methods
We conducted two independent, multicenter, double-blind, placebo-controlled studies in men undergoing unilateral inguinal hernioplasty by open laparotomy to evaluate the safety and efficacy of XaraColl at different doses (100 mg and 200 mg of bupivacaine hydrochloride; study 1 and 2, respectively). Enrolled patients (50 in study 1 and 53 in study 2) were randomized to receive active or placebo implants in a 1:1 ratio. Postoperative pain intensity and use of supplementary opioid medication were recorded through 72 hours. Safety was assessed through 30 days. The principal efficacy variables were the summed pain intensity (SPI), total use of opioid analgesia (TOpA), and an integrated endpoint (I-SPI-TOpA). Each variable was analyzed at 24, 48, and 72 hours after implantation. A pooled analysis of both studies was also performed retrospectively.
Results
Through 24 and 48 hours, XaraColl-treated patients experienced significantly less pain in study 1 (P < 0.001 and P = 0.012, respectively) whereas they took significantly less opioid analgesia in study 2 (P = 0.004 and P = 0.042, respectively). Over the same time intervals in the pooled analysis, treated patients experienced both significantly less pain (P < 0.001 and P = 0.006, respectively) and took significantly less opioid analgesia (P = 0.001 and P = 0.024, respectively). The I-SPI-TOpA endpoint that combined both SPI and TOpA demonstrated a significant treatment effect through 72 hours in the pooled analysis (P = 0.021).
Conclusion
XaraColl offers great potential for improving the management of postoperative pain and warrants further investigation in definitive clinical trials.
Keywords:
Introduction
Despite widely accepted treatment standards and guidelines, postoperative pain continues to be undermanaged.Citation1 In a survey of 250 postoperative patients, the most common concern identified was postoperative pain.Citation2 While opioid analgesics are the mainstay of treatment, there are well established risks associated with their use including sedation, nausea, vomiting, pruritus, respiratory depression, and immunosuppression.Citation1 Therefore, new interventions to manage postoperative pain better and reduce the dependence on opioid analgesics are needed.
XaraColl® (Innocoll Technologies, Athlone, Ireland) is a biodegradable and fully resorbable collagen matrix impregnated with the local anesthetic, bupivacaine, which is under development for postoperative analgesia. The product is implanted during a surgical procedure and releases bupivacaine for local, sustained action at the site(s) of trauma, while maintaining low systemic levels well below the drug’s neurotoxicity and cardiotoxicity thresholds. Because XaraColl starts releasing bupivacaine immediately, the patient is already benefiting from its analgesic effect upon recovery from their general anesthesia. However, this also poses some particular challenges when undertaking a clinical evaluation.
The scientific literature reveals two main approaches for assessing the efficacy of postoperative analgesics. The overwhelming majority of published methods are based either on the patient’s need for supplementary analgesia to control their pain adequately, or on the patient’s self-reporting of pain using a validated assessment tool, such as a visual analog scale or numeric rating scale.Citation3 However, both types of approach are known to suffer limitations. Concerns about toxicity or fear of addiction mean that some patients will opt to suffer more pain rather take adequate doses of postoperative analgesia (particularly opioids), whereas others will choose to dose more liberally.Citation4 Furthermore, the fact that patients must be permitted access to supplementary analgesia confounds the effect of the test drug when assessing their pain.
Silverman et al recognized these limitations and concluded that an integrated outcome variable that takes both pain intensity and use of supplementary (or “rescue”) analgesia into account should serve as the primary endpoint for assessing efficacy.Citation5 They developed a method by which ordinal ranks of pain score and use of rescue analgesia were expressed as percent differences from the mean ranks, and then summed both of these to generate a single, integrated variable that weighted the patient’s pain score and use of rescue analgesia equally. By integrating both types of traditional endpoints in this manner, a patient is appropriately penalized whether choosing to suffer more pain or take higher doses of rescue analgesia. However, because Silverman et al only considered pain intensity measured at a single time point, we have further adapted their method to manage multiple time points for analysis over different postoperative time intervals.
Another study design challenge specifically associated with perioperative implants is the lack of opportunity to assess a patient’s baseline (ie, untreated) pain or to perform any kind of crossover study, as is often used in studies of chronic pain. This makes it impossible to observe a treatment effect for any individual patient and, given the expected level of intersubject variability in pain perception, can limit the reliability of pilot clinical evaluations when sample sizes tend to be modest. For this reason, we also conducted a retrospective analysis by pooling the datasets from two similarly designed clinical studies.
Materials and methods
We conducted two independent, multicenter, randomized, single-dose, double-blind, placebo-controlled, pilot studies to evaluate the safety and analgesic effect of implanting two XaraColl implants in men undergoing inguinal hernioplasty. In the first study (study 1, NCT00626886), the implants each contained 50 mg bupivacaine hydrochloride for a total dose of 100 mg, and in the second study (study 2, NCT01220024), the XaraColl implants each contained 100 mg bupivacaine hydrochloride for a total dose of 200 mg. Both studies were conducted in the US at eight centers for study 1 and five centers for study 2. They were approved by an institutional review board, either centrally (Western Institutional Review Board, Olympia, WA) or locally at the trial center, and performed in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines.
Eligible patients included men at least 18 years of age who were generally healthy and scheduled for a unilateral inguinal hernioplasty by open laparotomy. Patients were excluded if they were scheduled for a bilateral inguinal hernioplasty requiring more than one incision, if they had undergone previous hernia repair on the same side, or if they had any concomitant illness that would significantly increase their surgical risk or make it difficult to complete the assessments required. We also excluded patients who were being treated for concomitant illness with agents that could affect the analgesic response, such as central alpha agents, neuroleptic agents, and other antipsychotic agents, monoamine oxidase inhibitors, or systemic corticosteroids. Patients who were deemed eligible provided written informed consent and underwent screening procedures, including a physical examination and routine laboratory tests up to 6 weeks before surgery. On the day of surgery, patients underwent confirmatory safety assessments and those who continued to meet the study entry criteria were randomized in a 1:1 ratio to the XaraColl treatment or matching placebo control group. An independent statistician created a randomization schedule that was used by the contractor (Almac, Craigavon, Northern Ireland) responsible for packaging and labeling the clinical supplies, to create blinded, numbered study kits. The kits were designed with a two-part label: one part remained affixed to the kit box, and the other was affixed to the patient’s case report form. The label contained a scratch-off section that would reveal the treatment assignment in the event the blind needed to be broken, such as for a serious adverse event. The sites were instructed to dispense the kits in the numeric order in which they were received. The investigators, patients, and study staff were blinded to treatment assignment throughout each study and the blind was not broken until the databases were closed and locked.
Surgery was conducted under general anesthesia that allowed short-acting agents, such as propofol, midazolam, and fentanyl. The use of epidural anesthesia or local anesthetic infiltrations was prohibited. Two XaraColl or matching placebo implants were administered to each patient; the first was positioned into the abdominal wall repair with the reinforcement mesh placed over it and the second was implanted directly below the skin incision prior to wound closure. The time at which the first implant was administered was defined as time 0.
Following surgery, patients were observed and evaluated by hospital staff for a minimum of 6 hours. Immediate postoperative pain was treated with intravenous morphine at incremental doses of 1–2 mg as needed to achieve pain control. Once patients could tolerate oral medication, they were provided with immediate release oral morphine tablets as rescue analgesia and instructed to take only if necessary for breakthrough pain. After discharge, patients returned to the study site at 72 hours after surgery for their final efficacy assessments and follow-up visit. Hospital staff contacted patients by telephone approximately one week and one month after surgery to inquire about their surgical wound, use of concomitant medications, any adverse events, and their general well-being.
Patients assessed their pain intensity after aggravated movement (defined as cough) using a horizontal 100 mm visual analog scale with the left anchor (0 mm) labeled “no pain” and the right anchor (100 mm) labeled “worst pain imaginable.” In study 1, these pain assessments were scheduled at approximately 1, 1.5, 2, 3, 4, 5, 6, 24, 48, and 72 hours after time 0 and, in study 2, they were scheduled at approximately 1, 2, 4, 6, 8, 10, 12, 24, 48, and 72 hours after time 0. In both studies, the use of all opioid analgesia administered from time 0 through 72 hours was recorded and converted to intravenous morphine equivalent using a published conversion table.Citation6
We collected safety data, including physical findings, vital signs, and laboratory assessments, at scheduled intervals and recorded all adverse events and serious adverse events throughout the study duration. An adverse event was any clinically unfavorable and unintended sign (including abnormal laboratory findings), symptom, or disease whether or not it was causally related to the treatment. A serious adverse event was any adverse event that resulted in death, was life-threatening, required inpatient hospitalization or prolongation of existing hospitalization, resulted in permanent disability/incapacity, or was an important medical event. We designated each adverse event based on clinical severity as either “mild” (causes no limitation of usual activities), “moderate” (causes some limitation of usual activities), or “severe” (prevents or severely limits usual activities). We also assessed its expectedness and its relationship to treatment as either “definitely related”, “probably related”, “unlikely related”, or “not related”. Finally, the outcome of each adverse event at study completion was assessed and reported as either “recovered”, “resolved with sequelae”, “ongoing”, “death”, “other”, or “unknown”.
Endpoints and statistical methods
The principal efficacy variables analyzed for each study were SPI, TOpA, and I-SPI-TOpA, based upon the method described by Silverman et al.Citation5 Each variable was analyzed at three time intervals, ie, 24, 48, and 72 hours after time 0, to yield nine efficacy endpoints of particular interest.
The SPI was defined and calculated as the trapezoidal area under the visual analog scale curve from one hour through to 24, 48, or 72 hours after time 0, regardless of the actual time the assessments were recorded. Any pain intensity assessments missing from those scheduled were imputed according to the following prospective rules:
Any missing pain intensity assessments before the first observed assessment were imputed with the patient’s worst observation (ie, their highest recorded visual analog scale score).
All other missing pain intensity assessments were imputed using the patient’s last observation (ie, the observation prior to that missing); however, in the event that a patient had to be terminated early from the study and the implant removed, the patient’s worst observation would have been imputed for all missing data.
The I-SPI-TOpA parameter was calculated for each time interval as follows:
All patients in the combined treatment groups were ranked according to their SPI value; if a tie occurred, the average rank was applied to the tied observations as if there were no ties.
The mean rank of the combined treatment groups was calculated.
The mean rank was subtracted from each patient’s SPI rank and expressed as a percentage of the mean rank (“% difference”); this yields positive values for patients having a higher SPI than the median and negative pain scores for patients having a lower SPI.
The above steps were repeated for TOpA.
For each patient, the percent differences derived for the SPI and TOpA were summed to generate the summated percent difference, which we termed I-SPI-TOpA.
The planned enrollment for study 1 and 2 was 52 and 50 patients, respectively. For study 1, we calculated that 26 patients in each group provided 83% power to detect an estimated 5 mg reduction in TOpA, assuming a common standard deviation of 6 mg and using a two-sided t-test at the 0.05 significance level. For study 2, we calculated that 25 patients in each group provided 81% power to detect an estimated 1237 mm · hour reduction in SPI, assuming a common standard deviation of 1546 mm · hour and using a two-sided t-test at the 0.05 significance level. The estimated reduction in SPI and common standard deviation were based on actual observed values from study 1.
Efficacy analyses for both studies were based on the predefined intent-to-treat population, which consisted of all randomized patients who had at least two visual analog scale pain intensity scores. All pairwise comparisons were two-sided and performed using one-way analysis of variance (ANOVA) models with treatment as the main effect. Summary statistics and the P-value from the ANOVA model were computed for each efficacy variable using SAS® Software version 8.2 or higher (SAS Institute Inc, Cary, NC). P-values ≤ 0.05 were considered statistically significant and P-values > 0.05 but ≤ 0.10 were considered indicative of a trend towards statistical significance.
To maximize the statistical power available, we also conducted the same set of analyses after retrospectively pooling the data from both intent-to-treat populations. For each of the nine efficacy variables of interest, we fitted an ANOVA model that included fixed effects for treatment (active or placebo), study/dose (study 1, 100 mg or study 2, 200 mg), and treatment-by-study interaction. We assumed the pooled analysis was statistically valid for a given efficacy variable if there was no evidence of an interaction at the 0.05 significance level.
Safety evaluations for both studies were based on all randomized patients and included summary reports for the incidence and severity of adverse events, their relationship to the treatment, and any clinically significant changes or abnormalities in the patient’s physical examination, vital signs, or laboratory results.
Results
From March 2008 to January 2009, we enrolled 53 patients in study 1 (24 were randomized to XaraColl and 29 to placebo) and, from December 2010 to May 2011, we enrolled 50 patients in study 2 (25 were randomized to both XaraColl and placebo). The disposition of all enrolled patients is summarized in and the patient demographics, which were similar across treatment groups and studies, are presented in . Two patients in study 2 (one in each treatment group) were unable to be contacted for the one-week and one-month telephone call, and therefore were considered lost to follow-up for safety after the 72-hour assessment. One patient in study 2 (randomized to the control group) was eliminated from the intent-to-treat population for failing to record any pain assessments.
Figure 1 CONSORT flow diagram for study 1 and study 2.
Abbreviation: ITT, intent-to-treat population.
Table 1 Patient demographics
Efficacy
Results of the analyses for the three principal efficacy endpoints (SPI, TOpA and I-SPI-TOpA) as performed through 24, 48, and 72 hours for each of the three datasets (study 1, study 2, and pooled) are summarized in . SPI, TOpA, and I-SPI-TOpA are also displayed graphically in -, respectively. In the pooled analysis, the treatment-by-study interactions were all nonsignificant (P ≥ 0.124), and hence pooling was considered to be valid for all the efficacy variables analyzed.
Figure 2 SPI for study 1, study 2, and pooled dataset through 24, 48, and 72 hours.
Abbreviation: SPI, summed pain intensity.
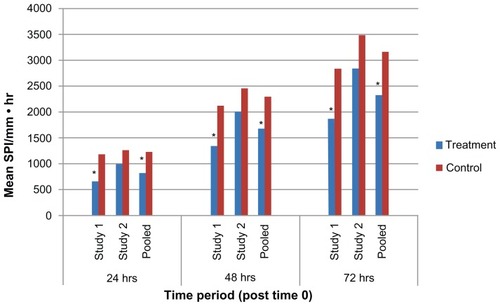
Figure 3 TOpA for study 1, study 2, and pooled dataset through 24, 48, and 72 hours.
Abbreviation: TOpA, total opioid analgesia.
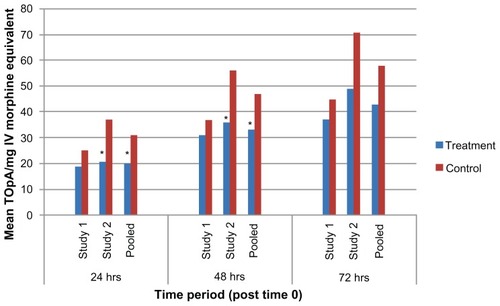
Figure 4 I-SPI-TOpA through 24, 48, and 72 hours, study 1, study 2, and pooled dataset.
Abbreviations: I-SPI-TOpA, integrated SPI and TOpA variable; SPI, summed pain intensity; TOpA, total use of opioid analgesia.
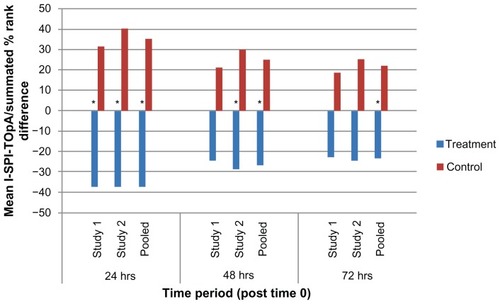
Table 2 Results for SPI, TOpA, and I-SPI-TOpA through 24, 48, and 72 hours for study 1, study 2, and pooled dataset
Over the first 24 hours following surgery, the SPI demonstrated that patients treated with XaraColl had significantly less pain in study 1 and the pooled dataset (P < 0.001), and trended towards significance in study 2 (P = 0.080). Over the same time period, treated patients also took significantly less opioid medication in study 2 (P = 0.004) and the pooled dataset (P = 0.001). However, the corresponding TOpA analysis was not statistically significant in study 1. This may have been because a patient in the treatment group reportedly dosed his opioid medication as maximally allowed, rather than as needed for pain control per instructions. Despite this patient, all treated patients in the pooled dataset still took 37% less opioid analgesia than the control patients, while also experiencing 32% less pain. Importantly, when taking both pain and use of rescue medication into account, the I-SPI-TOpA analyses showed a statistically significant treatment effect for both individual studies and was highly significant for the pooled dataset (P = 0.013, P = 0.005, and P < 0.0001 for study 1, 2, and pooled, respectively).
Observations from the first 48 hours post-surgery were similar to those for the first 24 hours. XaraColl-treated patients had significantly less pain in study 1 (P = 0.012) and the pooled dataset (P = 0.006), but not in study 2. With respect to rescue analgesia, patients in the treated group took significantly less opioid medication in study 2 (P = 0.042) and the pooled dataset (P = 0.024), but not in study 1. On further examining the reported incidence of nausea in study 2, we observed a 52% reduction in the treatment group (16.0%, 4/25 patients) compared with the control group (33.3%, 8/24 patients). We also found that the nausea events reported by XaraColl-treated patients were generally less severe and of shorter duration, despite more patients in the control group having received antiemetic medication for nausea treatment and/or prophylaxis. Through 48 hours, the combined I-SPI-TOpA endpoint continued to show a statistically significant treatment effect in study 2 (P = 0.040) and the pooled dataset (P = 0.008), with a trend towards significance in study 1 (P = 0.097).
Findings from the full 72 hours after surgery continued to show evidence of a XaraColl treatment effect, particularly the analysis of pooled data. The means of SPI and TOpA were both reduced by 26% compared with control, although this reduction was only statistically significant for pain (P = 0.014) and not opioid use. The pooled data also revealed a statistically significant reduction in the combined I-SPI-TOpA endpoint (P = 0.021). With regard to the individual studies, only the reduction in SPI for study 1 was statistically significant (P = 0.030). However, for study 2, the reductions in TOpA and I-SPI-TOpA both showed a trend toward significance (P = 0.094 and 0.078, respectively).
Taken together, these analyses of the different datasets demonstrate some striking features. Firstly, it seems that the assessment of pain was a more sensitive endpoint in study 1, whereas patient use of rescue analgesia was more discriminatory between treatment groups in study 2. This could possibly be attributed to the different doses of XaraColl in these studies. However, it is not apparent why that should be the case and, notably, the pooled analysis revealed no evidence of a significant treatment-by-study interaction. Therefore, a more likely explanation is differing patient attitudes to opioid medication when managing their postoperative pain. If this assertion is correct, then our studies offer a compelling reason for preferring an endpoint that integrates both pain and use of rescue analgesia into a single variable, such as I-SPI-TOpA.
Secondly, our research illustrates the power of pooling both datasets. For all nine endpoints of interest, the XaraColl treatment effect was more significant in the pooled analysis than in either individual study. Although unsurprising, this finding confirms the value of an increased sample size in situations where an analgesic acts before baseline pain can be assessed.
Finally, and most importantly, the results of the I-SPI-TOpA analyses provide strong evidence that XaraColl significantly reduces the combination of both pain and use of opioid medication over at least 48 hours and, based on the pooled analysis, up to 72 hours postoperatively.
Safety
In study 1, the most common adverse events (occurring in ≥10% of patients) in either treatment group were constipation, nausea, and headache. Most adverse events were considered mild in severity. One patient in the placebo group reported an adverse event of nausea that was considered treatment-related, but was not considered either serious or severe. Two serious adverse events, one in each treatment group (hypotension in the XaraColl group and hiccups in the placebo group) were reported, but neither was considered treatment-related. No deaths or discontinuations due to an adverse event were reported.
In study 2, the most common adverse events (ie, those occurring in ≥5% of patients in either treatment group) were constipation, nausea, scrotal swelling, vomiting, dizziness, chills, pyrexia, and incision site infection. All were considered mild (25 patients, 50.0%) or moderate (7 patients, 14.0%); none were considered severe or treatment-related. No deaths, serious adverse events, or discontinuations due to an adverse event were reported. These safety data confirm that XaraColl is safe and well tolerated in men undergoing hernioplasty up to doses of 200 mg bupivacaine hydrochloride.
Conclusion
Clinical evaluation of perioperative implants aimed at providing postoperative analgesia is complicated by concomitant use of systemically acting analgesics and inability to measure baseline pain, particularly in pilot studies with modest sample sizes. Therefore, we have adapted the methodology first proposed by Silverman et alCitation5 to create an integrated efficacy endpoint, I-SPI-TOpA, that combines the patient’s SPI and TOpA, and appears to address the limitations of using either endpoint alone. Through use of I-SPI-TOpA in addition to those traditional endpoints, we have demonstrated that XaraColl, a perioperative bupivacaine-collagen implant, significantly reduces pain and use of opioid medication over at least 48 hours after hernioplasty, as based on two independent pilot studies performed at safe and well tolerated doses of bupivacaine. Furthermore, a pooled analysis of both studies indicates a statistically significant treatment effect over 72 hours after surgery. These findings suggest that XaraColl offers great potential for the management of postoperative pain and warrants definitive clinical trials with larger sample sizes.
Acknowledgments
We thank the following investigators and their study staff who enrolled patients in the studies: Todd Gerkin (Vital Research), Mark Hanley (East Coast Clinical Research Group), Laurence Kam (Memorial Hermann Memorial City Hospital), Johanna Knust (Northwest Surgeons/HealthFirst Medical Group), Tim Melson (Helen Keller Hospital and Eliza Coffee Memorial Hospital), Harold Minkowitz (Memorial Hermann Memorial City Medical Center), Dennis Riff (Advanced Clinical Research), Paul Siami (Welborn Clinic Research Center), Steven Wininger (Precision Trials), Peter Winkle (Advanced Clinical Research Institute), and Jonathan Yunis (Sarasota Memorial Hospital).
Disclosure
This study was fully funded by Innocoll Technologies, and was performed by Premier Research, which recruited and monitored the sites, managed the clinical databases, analyzed the data, and wrote the study reports. HSM and PW received research funding from Innocoll Technologies. MJ and SLC are paid consultants to Innocoll Technologies.
References
- WuCLRajaSNTreatment of acute postoperative painLancet20113772215222521704871
- ApfelbaumJLChenCMehtaSSGanTJPostoperative pain experience: results from a national survey suggest postoperative pain continues to be undermanagedAnesth Analg200397253454012873949
- HjermstadMJFayersPMHaugenDFStudies comparing numerical rating scales, verbal rating scales, and visual analogue scales for assessment of pain intensity in adults: a systematic literature reviewJ Pain Symptom Manage20114161073109321621130
- Wilder-SmithCHSchulerLPostoperative analgesia: pain by choice? The influence of patient attitudes and patient educationPain19925032572621454382
- SilvermanDGO’ConnorTZBrullSJIntegrated assessment of pain scores and rescue morphine use during studies of analgesic efficacyAnesth Analg19937711681708317727
- GordonDBStevensonKKGriffieJMuchkaSRappCFord-RobertsKOpioid equianalgesic calculationsJ Palliat Med19992220921815859817