Abstract
Objective
To characterize the steady-state pharmacokinetic profile of hydromorphone extended-release (ER) in patients with chronic pain taking concomitant medications.
Methods
This open-label repeat-dose study enrolled 22 patients (mean age, 51.4 years; 81.8% female). All patients were receiving at least one concomitant medication; 86.4% were receiving at least two concomitant medications and 81.8% were receiving at least three. Patients receiving a stable dose of an opioid were converted to hydromorphone ER at a 5:1 ratio (morphine equivalent:hydromorphone). The dose was titrated to adequate analgesia over 3–14 days and stabilized between 8–48 mg. Oral morphine immediate-release was permitted for breakthrough pain. Area under the concentration–time curve from 0–24 hours (AUC0–24), maximum plasma concentration (Cmax), trough plasma concentration (Cmin), average plasma concentration (Cavg), and degree of fluctuation (100 × [(Cmax − Cmin) ÷ Cavg]) were calculated based on data from 14 patients.
Results
Dose-normalized to 16 mg, mean pharmacokinetic parameter values were: AUC0–24, 41.1 ng · h/mL; Cmax, 2.6 ng/mL; Cmin, 1.1 ng/mL; Cavg, 1.7 ng/mL; and the degree of fluctuation was 99.6%. The pharmacokinetic profile of hydromorphone ER was linear and consistent with dose proportionality. Mean pain intensity difference scores showed statistically significant improvement from 2–21 hours after dosing. Sixteen (72.7%) patients reported at least one adverse event (AE). The most common were constipation (31.8%), headache (22.7%), and vomiting (13.6%). One patient discontinued treatment due to vomiting. No deaths, serious AEs, or unexpected AEs occurred.
Conclusion
These findings replicate and extend the steady-state pharmacokinetic profile of hydromorphone ER, previously characterized in healthy volunteers, to a population of chronic pain patients taking numerous concomitant medications.
Introduction
“Chronic pain” is defined as pain that extends beyond the expected period of healing and persists for ≥3 months.Citation1 Between 1999 and 2002, the Centers for Disease Control and Prevention estimated that 26% of US adults (52 million people) reported pain that lasted for more than 24 hours in the previous month.Citation2,Citation3 Of those respondents who reported pain, approximately 56% (29 million people) reported having pain that lasted for 3 months or longer.Citation2,Citation3 Chronic pain is associated with deleterious effects on physical, mental, and social functioning, and significantly affects quality of life.Citation4,Citation5
A number of pharmacologic options are available for the management of chronic pain, including nonopioid and opioid analgesics. Patients with moderate to severe chronic pain that affects functioning or quality of life may benefit from a trial of chronic opioid therapy.Citation6 Current guidelines published by the American Pain Society and the American Academy of Pain Medicine support the use of extended-release (ER) opioids and recognize their putative advantages over short-acting immediate-release (IR) formulations.Citation6 Once- or twice-daily dosing of ER opioids may provide more consistent analgesia and facilitate patient adherence to treatment regimens.Citation7 Additionally, less frequent dosing and more stable and consistent drug plasma levels may decrease end-of-dose failure, “clock watching” between doses, and the “wind-up” phenomenon.Citation8,Citation9
The semisynthetic opioid analgesic hydromorphone has been used extensively in the treatment of chronic pain conditionsCitation10,Citation11 and is approximately four to eight times more potent than morphine.Citation12–Citation16 Hydromorphone is unlikely to be associated with significant drug–drug interactions involving cytochrome P450 (CYP450) because it is metabolized primarily via glucuronidation.Citation17 Hydromorphone has been available for some time as an IR formulation; however, chronic pain may be better managed in some patients with the long-lasting analgesic effects of the oral ER formulation of hydromorphone.Citation18
Hydromorphone ER tablets produce sustained, monophasic delivery of hydromorphone over a 24-hour period, with dose-proportional pharmacokinetics.Citation18,Citation19 In previous studies in healthy volunteers, steady state was achieved by Day 4 in subjects taking 16 mg of once-daily hydromorphone ER.Citation18,Citation20 Hydromorphone ER maintains steady-state plasma drug concentrations within the same range as the IR formulation, with less mean peak-to-trough fluctuation (60.5% vs 172.0%).Citation20
Although the pharmacokinetic profile of hydromorphone ER has been established in healthy subjects, the pharmacokinetics of opioid formulations may differ in chronic pain patients, who are often older, have comorbid conditions, and take concomitant medications for these conditions. In a review of the pharmacokinetics of oral morphine formulations, for example, the maximal plasma concentration was higher and more variable in patients versus healthy subjects – results that have important safety implications.Citation21 An assessment of the pharmacokinetic profile of hydromorphone ER in chronic pain patients may provide a better appraisal of the real-world clinical pharmacology of this formulation. The objective of this open-label repeat-dose study was to characterize the steady-state pharmacokinetic profile of hydromorphone ER in chronic pain patients who are taking concomitant medications to manage comorbid conditions.
Methods
Study population
Men and women aged ≥ 18 years with chronic cancer or non-cancer pain receiving strong oral or transdermal opioid analgesics daily were enrolled. Patients eligible for advancement of therapy to Step 3 on the World Health Organization analgesic ladder were also included. Eligible patients required the opioid equivalent of ≥32 mg but ≤300 mg of oral morphine sulfate (exclusive of breakthrough pain medication) every 24 hours to manage their chronic cancer or non-cancer pain.
The following patients were excluded: women who were pregnant or breastfeeding (patients were required to submit a negative pregnancy test prior to administration of study drug); patients intolerant of or hypersensitive to hydromorphone or other opioid agonists; patients who had dysphagia or were unable to swallow tablets; patients currently taking an investigational drug or who had received an investigational drug within 30 days before study entry; patients with more than three episodes of vomiting per day within the 3 days before the start of the study and patients with intractable nausea; patients with acute abdominal conditions that could be obscured by opioids; patients with any significant central nervous system disorder (eg, seizure, stroke, or intracranial lesion) within the 6 months before the start of the study and patients with cognitive disorders; patients at risk for serious decreases in blood pressure following administration of an opioid, constipation (defined as having no bowel movement or having bowel obstruction due to impaction within the 5 days prior to study start), significant central nervous system disorders, impaired hematological, renal, or hepatic function, respiratory compromise, or any gastrointestinal disorder (eg, gastrointestinal narrowing) that could affect the absorption or transit of orally administered drugs; patients who were known active drug or alcohol abusers; and patients who had undergone drug or alcohol detoxification within 1 year of the start of the study.
Study design
This was an open-label repeat-dose study designed to establish the pharmacokinetic profile of OROS® hydromorphone ER (Exalgo®, Mallinckrodt Brand Pharmaceuticals, Hazelwood, MO) in patients with chronic pain conditions. The study was initiated on March 31, 1999, and completed on August 9, 1999, as part of a lengthy clinical development and regulatory review process that culminated in approval of the formulation by the US Food and Drug Administration in March 2010.
The study design is shown in . At Visit 1, each patient received a diary in which to record daily opioid medication use and pain relief ratings for the duration of the study. The study was conducted in accordance with the International Conference on Harmonisation good clinical practice guidelines and the principles of the Declaration of Helsinki. The protocol and all amendments were reviewed by the following independent institutional review boards: Southern Institutional Review Board (Miami, FL), Peninsular Testing Corporation Protocol Review Committee (Miami, FL), and Scripps Memorial Hospital (La Jolla, CA). Written informed consent was obtained from all study participants.
Figure 1 Study design.
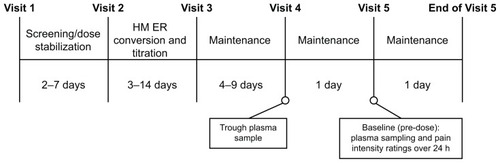
Following stabilization on their prior oral or transdermal opioid, all patients were converted to hydromorphone ER at a ratio of 5:1 (morphine equivalent:hydromorphone) at Visit 2. There was no placebo or other comparator group. The dose of hydromorphone ER was titrated to adequate analgesia during a period of 3–14 days and potentially stabilized using the 8 mg, 16 mg, 32 mg, and 64 mg tablets alone or in a dosing combination if the dose was different than the available strengths, as necessary. A patient’s dose was considered stabilized when the total daily dosage of hydromorphone ER remained unchanged for 2 consecutive days, with a maximum of three rescue medication doses per day. If, after 2 days of stable therapy, more than three doses of rescue medication were required in a 24-hour period, upward titration was considered. The hydromorphone ER dose could not be titrated more frequently than every 2 days. If patients could not achieve a stable dose of hydromorphone ER for 2 consecutive days (requiring no more than three doses of rescue medication), after 14 days of therapy, they were discontinued from the study.
Patients whose dose was stabilized were entered into the 4- to 10-day maintenance phase of the study. At Visit 4, occurring after a minimum of 4 days of therapy with hydromorphone ER at a constant daily dose, patients returned to the study clinic for an initial trough blood sample taken immediately prior to the usual daily dose of hydromorphone ER. The next morning (Visit 5), patients were required to return to the study clinic and remain for a 24-hour period. During this time, baseline plasma sample and pain intensity ratings were obtained prior to dosing and pharmacokinetic and pharmacodynamic assessments were conducted.
Study medications
At the start of the maintenance phase, patients received a 10-day supply of open-label hydromorphone ER; a stable dose could be achieved by a combination of 8 mg, 16 mg, 32 mg, and 64 mg tablets. During the maintenance phase, patients were instructed to take their medication at approximately the same time each day with approximately 240 mL (8 oz) of water. Additionally, patients were instructed not to chew, divide, or crush the hydromorphone ER tablet. Use of oral morphine sulfate IR (15 mg or 30 mg tablets) was permitted as rescue medication as needed for the management of breakthrough pain.
Plasma sampling
At Visit 5, blood samples were taken before dosing with hydromorphone ER and at 1 hour and 2, 4, 6, 8, 10, 12, 15, 18, 21, and 24 hours after dosing. A liquid chromatography–mass spectrometry method (SCIEX API-III “Plus” LC/MS/MS equipped with a short high-performance liquid chromatography column) (CEDRA Corporation, Austin, TX) was used to measure hydromorphone plasma concentrations, validated for a range of 0.05–10.0 ng/mL, which was based on the analysis of 1.00 mL of human plasma. After extracting plasma containing hydromorphone and the tri-deuterated internal standard with ethyl acetate, the organic layer was removed and evaporated to dryness. This extract was then reconstituted and an aliquot was injected onto a liquid chromatograph–mass spectrometer equipped with a short high-performance liquid chromatography column. Peak areas of the hydromorphone and the internal standard product ions were measured against each other. Quantitation was performed using 1/x-weighted linear least squares regression generated from calibration standards spiked immediately before each run.
Pharmacokinetic assessments
The primary objective of this analysis was to characterize the steady-state pharmacokinetic profile of hydromorphone ER by evaluating several pharmacokinetic parameters. Non-compartmental analysis was performed with WinNonlin® software (Professional Network version 2.1) (Pharsight, Mountain View, CA) to determine the steady-state pharmacokinetic profile of hydromorphone ER. The pharmacokinetic parameters of maximum plasma concentration (Cmax), trough plasma concentration (Cmin), average plasma concentration (Cavg), and area under the curve from 0–24 hours (AUC0–24) were determined from the concentration–time data (dose-normalized to 16 mg and non-normalized) for each patient. Time to maximum plasma concentration (Tmax), degree of fluctuation, and time to trough plasma concentration (Tmin) were calculated as non-normalized parameters.
Pharmacodynamic assessments
The secondary objective of this analysis was to determine the pharmacodynamic effects of administration of hydromorphone ER. The primary pharmacodynamic efficacy measure was pain intensity, which was assessed at Visit 5 using item 6 on the Brief Pain Inventory Short Form,Citation22 “pain right now.” Patients were required to record a pain intensity score in their diaries based on an 11-point scale (0 = no pain and 10 = pain as bad as you could imagine) immediately prior to having each blood sample taken during the 24-hour period. An increase in pain intensity score indicated that pain was worsening and a decrease in pain intensity score indicated that pain was improving. The measurement of pain intensity difference (PID) from baseline was calculated at each time point. A small or negative value for PID from baseline indicated that pain relief was not being achieved or that pain was worsening while a larger value for PID indicated pain relief. Individual and mean area under the effect curve (AUEC) for each effect (pain intensity and PID) were calculated using the trapezoidal method.
Two secondary pharmacodynamic efficacy measures were also evaluated. Pain relief (0 = no relief, 1 = poor relief, 2 = moderate relief, 3 = good relief, 4 = complete relief) was assessed according to diary-based pain relief scores. Additionally, patient and investigator global evaluation (5-point categorical measure of effectiveness of study medication: 1 = poor, 2 = fair, 3 = good, 4 = very good, 5 = excellent) was conducted at the conclusion of Visit 5. Pain relief scores and patient and investigator global evaluation scores were tabulated and summarized.
Adverse events (AEs)
AEs were recorded at each visit and coded using the Hoechst Adverse Reaction Terminology System dictionary.Citation23 AEs were rated by the investigator as mild, moderate, or severe. “Mild AEs” were defined as AEs that did not limit patients’ usual activities but may have caused slight discomfort. “Moderate AEs” were defined as AEs that limited patients’ usual activities and may have caused significant discomfort. “Severe AEs” were defined as AEs that prevented patients from carrying out their usual activities and may have caused intolerable discomfort or pain. AEs were also judged by the investigator to be “unrelated,” “unlikely,” “possibly,” or “probably” related to the study drug.
Potential AEs that were classified as serious included death, any AE that was considered life-threatening or caused hospitalization, any AE that caused a significant disability or incapacity or a congenital anomaly/birth defect, or any medical event or situation that jeopardized the patient or required medical or surgical intervention to prevent one of the outcomes listed in this definition.
Statistical analyses
For the non-compartmental pharmacokinetic analysis, standard methods were used to determine the summary statistics (mean, median, standard deviation [SD], coefficient of variation, minimum, and maximum) of dose-normalized (to 16 mg) and non-normalized pharmacokinetic parameters. Simple linear regression analyses were performed on Cmax, Cmin, Cavg, and AUC0–24 as a function of hydromorphone ER dose to investigate linearity of hydromorphone ER pharmacokinetics. For the area under the curve of pain intensity and PID, summary statistics were determined using standard methods. A post-hoc analysis measuring the time spent ≥ 50% Cmax was performed using data from 14 patients. The time ≥ 50% Cmax was the total time during the 24-hour dosing interval that the plasma concentration was ≥50% of Cmax and included linear interpolation between plasma concentration–time points that crossed the 50% threshold. AE data were displayed, tabulated, and summarized in a descriptive manner.
Results
Patients
Initially, 22 patients were enrolled and all patients received at least one dose of hydromorphone ER. During the titration phase, two patients withdrew from the study because of protocol violations (one because of an unsanctioned dose increase and one because of history of dysphagia). One patient withdrew due to vomiting during the titration phase. During the maintenance phase, one patient discontinued treatment because of a discrepancy in study medication pill count. Eighteen patients completed the study; patient disposition is illustrated in . Of the 22 patients enrolled in the study, 18 (81.8%) were female and 20 (90.9%) were white, with a mean age of 51.4 years (range, 25–81 years). A majority (n = 19; 86.4%) presented with musculoskeletal pain; the remaining three patients (13.6%) presented with neuropathic pain. The overall mean (SD) pain intensity score at baseline for the 17 patients for whom complete data were available was 5.5 (2.1); the median pain intensity score was 6.0 ().
Figure 2 Patient disposition.
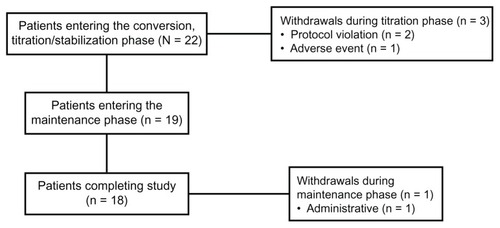
Table 1 Baseline demographics and characteristics
All patients included in this analysis took at least one concomitant medication while receiving hydromorphone ER. Nineteen (86.4%) patients took two or more concomitant medications, and 18 (81.8%) took three or more. Concomitant medications and medication classes are detailed in . The most common concomitant medications were hormone therapies, cardiovascular medications, and antidepressants.
Table 2 Concomitant medication use
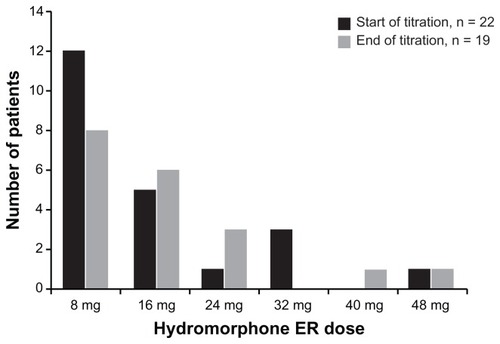
During the titration phase, 13 patients (59.1%) achieved a stabilized dose on their initial dose of hydromorphone ER without titration; these patients’ doses were stabilized on the initial 5:1 equi-analgesic conversion dose of hydromorphone ER. Five patients (22.7%) required one titration step and four patients (18%) required two steps to reach a stabilized dose of hydromorphone ER. Those patients who required titration (n = 9; 40.9%) required a median of 7.5 days to reach a stabilized dose of hydromorphone ER. A total of 19 of the 22 patients (86.3%) who entered the titration phase continued in the study. At the end of the titration phase, the dose of hydromorphone ER was stabilized at 8 mg for eight of these 19 patients (42.1%), 16 mg for six patients (31.6%), 24 mg for three patients (15.8%), 40 mg for one patient (5.3%), and 48 mg for one patient (5.3%) (). The mean (SD) dose of hydromorphone ER in patients entering the maintenance phase was 16.8 (11.3) mg.
Figure 3 Distribution of OROS® hydromorphone extended-release (ER) doses at the start and end of the titration phase.
Pharmacokinetic parameters
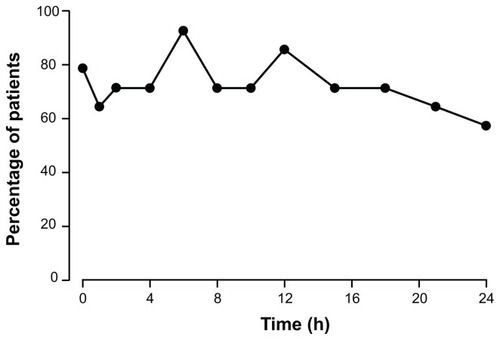
Mean pharmacokinetic parameters at steady state were calculated for data from 14 patients. The majority of patients had comparable hydromorphone plasma concentrations at both Visit 4 and Visit 5, indicating that steady state had been achieved. Pharmacokinetic data were dose-normalized to the 16 mg dose of hydromorphone ER. The mean (SD) Cmax, Cmin, and Cavg were 2.6 (0.8) ng/mL, 1.1 (0.4) ng/mL, and 1.7 (0.5) ng/mL, respectively. The mean (SD) AUC0–24 was 41.1 (12.7) ng · h/mL. After the administration of hydromorphone ER, the mean (SD) Tmax and Tmin were 8.1 (4.9) hours and 9.5 (8.5) hours, respectively. The dose-normalized concentration–time profile of hydromorphone ER at steady state is depicted in . Across the dose range studied, there was a significant linear relationship between the dose and AUC0–24. Dose proportionality was demonstrated across the doses studied (8–48 mg) (). In addition, dose proportionality was maintained after analyzing the data with the data of the one patient receiving 48 mg excluded. Under steady-state conditions, there was also a significant linear relationship (P < 0.05) between hydromorphone ER dose and Cmax, Cmin, Cavg, and AUC0–24 (data not shown). The mean (SD) degree of fluctuation (100 × [(Cmax − Cmin) ÷ Cavg]) was 99.6% (58.4). In a post-hoc analysis, the overall mean (SD) time spent ≥ 50% Cmax was 19.4 (4.3) hours. shows the percentage of patients in whom plasma levels were ≥50% Cmax at each time point during the 24-hour dosing period. At hour 6, for example, plasma concentrations ≥ 50% Cmax were recorded for nearly all (92.9%) patients. Additionally, at hour 24, plasma concentrations ≥ 50% Cmax were recorded for more than half (57.2%) of all patients.
Figure 4 Mean (standard deviation) dose-normalized (to 16 mg dose) hydromorphone plasma concentration–time profile following the administration of OROS® hydromorphone extended-release (n = 17).
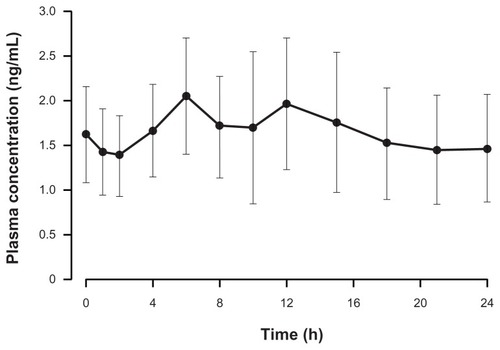
Figure 5 Relationship between hydromorphone area under the concentration–time curve from 0–24 hours (AUC0–24) and dose of OROS® hydromorphone extended-release (ER).
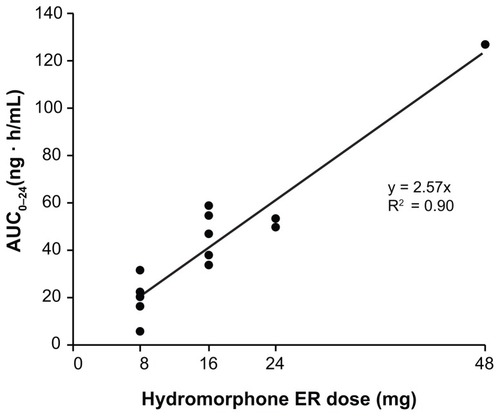
Figure 6 Percentage of patients with plasma concentrations ≥ 50% maximum plasma concentration at each time point after dosing with OROS® hydromorphone extended-release (n = 14).
Pharmacodynamic effects
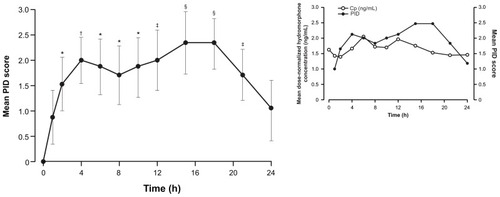
The pharmacodynamic effects of hydromorphone ER were evaluated in 17 patients. Hydromorphone ER was associated with a reduction in pain intensity, as demonstrated by an improvement in mean pain intensity (standard error of the mean [SEM]) scores across the 24-hour dosing interval. Mean (SEM) pain intensity scores improved from 5.5 (0.5) at baseline to 3.2 (0.6) at 18 hours after dosing and 4.5 (0.6) at 24 hours after dosing. Likewise, median pain intensity scores improved from 6.0 at baseline to 2.0 at 18 hours after dosing and 4.0 at 24 hours after dosing. Following administration of hydromorphone ER, mean differences in pain intensity (PID scores) were significantly different from baseline at all assessments except hour 1 and hour 24 ().
Figure 7 Mean (standard error of the mean) pain intensity difference (PID) scores over 24 hours at Visit 5 (n = 17).
The mean pain relief score (measured on a scale of 0–4) on the last day of the screening/stabilization phase was 1.86 (median, 2.0); the mean pain relief score improved to 2.32 and 2.58 by the last day of the titration and maintenance phases, respectively (median, 3.0 at end of maintenance). From screening to the last day of the titration phase, mean (SEM) change in pain relief score was 0.53 (0.24) (P < 0.05). From screening to the last day of maintenance, the mean (SEM) change in pain relief score was 0.67 (0.20) (P < 0.004). At the study termination visit, >90% of patients (n = 20) and investigators (n = 20) assigned hydromorphone ER a global evaluation score (measured on a scale of 1–5) of good to excellent, indicating effective pain relief.
A total of 13 patients (76.5%) used oral morphine as rescue medication to manage breakthrough pain during the 24-hour pharmacokinetic and pharmacodynamic assessments. Four (23.5%) patients did not require any rescue medication doses for breakthrough pain. At Visit 5, the average number of rescue medication doses (one dose = 15 mg) during the 24-hour period was 1.8. The mean (SD) morphine-equivalent total daily opioid dose (hydromorphone ER dose plus breakthrough medication) at the last day of maintenance was 125 (77.5) mg, compared with 87 (52.8) mg at the last day of stabilization prior to hydromorphone ER conversion. There was no correlation between the total daily dose of rescue medication and AUEC for pain intensity and PID (R2 = 0.0028 and 0.038, respectively). There was also no correlation between the total daily dose of rescue medication used and the hydromorphone ER dose (R2 = 0.006).
AEs
In total, 16 of 22 patients (72.7%) reported at least one AE during the study (). The most commonly reported AEs were constipation (n = 7; 31.8%), headache (n = 5; 22.7%), and vomiting (n = 3; 13.6%). In four cases of constipation, the relationship to the study drug was considered by the investigator to be possible; in one case, the relationship was considered probable. For two cases of vomiting, the relationship to the study drug was considered possible. Of the 37 AEs reported during the study, the investigators considered 24 (64.9%) and 13 (35.1%) mild or moderate in severity, respectively. No AEs were considered severe. One patient withdrew from the trial due to vomiting, which resolved following discontinuation of treatment with the study drug. This AE was rated by the investigators as mild and as being possibly related to hydromorphone ER. No serious AEs or deaths occurred during this study.
Table 3 Adverse events (AEs) reported by ≥5% of patients, by relationship to study drug
Discussion
This open-label repeat-dose study in patients with chronic pain taking a variety of concomitant medications was designed to confirm the steady-state pharmacokinetic profile of hydromorphone ER previously characterized in healthy subjects. The pharmacokinetic results indicated sustained steady-state plasma concentrations throughout the 24-hour dosing period after administration of hydromorphone ER and were consistent with results of previously published studies in healthy volunteers not taking concomitant medications.Citation20,Citation24 This population of patients with chronic pain, in which all patients took at least one concomitant medication, provides an accurate reflection of the real-world clinical setting. Results of this study confirm that hydromorphone ER produces a stable and consistent pharmacokinetic profile in patients with chronic pain taking a broad array of concomitant medications, suggesting that it may have a low risk for significant drug–drug interactions. Hydromorphone is not expected to have interactions with drugs metabolized through the cytochrome P450 system because it is primarily metabolized through glucuronidation.Citation25
The combination of the hydromorphone molecule and the controlled delivery kinetics of the formulation yielded consistent and sustained steady-state plasma concentrations throughout the 24-hour dosing period. Overall, hydromorphone ER doses were stabilized in 86.4% of patients (19 of 22), with doses ranging from 8–48 mg. When individual pharmacokinetic parameters were normalized to the 16 mg dose of hydromorphone ER, the mean Cmax, Cmin, Cavg, Tmax, and AUC0–24 pharmacokinetic results at steady state compared well with results obtained from previous studies in healthy volunteers.Citation20,Citation24 Furthermore, data from the AUC0–24 analysis are consistent with previous reports of dose proportionality across a broad range of doses (8–64 mg).Citation19,Citation26
The mean degree of peak–trough fluctuation with hydromorphone ER was 99.6%. In a study evaluating the pharmacokinetics of hydromorphone ER in healthy volunteers, the degree of fluctuation was somewhat lower (60.5%).Citation20 However, the degree of fluctuation reported in the current analysis is substantially lower than the 172.0% fluctuation reported for IR hydromorphone.Citation20 The consistency of around-the-clock dosing regimens can also be assessed by measuring the duration of time that a dosing regimen yields plasma concentrations that are ≥50% Cmax.Citation27 In an experimental acute-dose pain model evaluating the pharmacodynamics of hydromorphone ER versus IR hydromorphone, mean (SD) time spent ≥ 50% Cmax was 22.7 (8.2) hours compared with 1.1 (0.7) hours, respectively.Citation27 Investigators found a direct correlation between hydromorphone concentration and analgesia.Citation27 In the current study, the mean duration of concentrations ≥ 50% Cmax after administration of hydromorphone ER was 19.4 (4.3) hours during the 24-hour dosing period. Time spent ≥ 50% Cmax may emerge as an important consideration when selecting a treatment regimen for chronic opioid therapy.
Pharmacodynamic assessments in the current study indicated that the sustained plasma levels of hydromorphone were associated with decreased pain intensity throughout the 24-hour dose interval. As shown in the inset to , graphic displays of both dose-normalized plasma hydromorphone concentration and mean PID over 24 hours suggest a temporal association between changing plasma concentrations of hydromorphone and degree of analgesia. However, the design of the study did not allow the characterization of this correlation with any degree of certainty, given that the assessments were performed at steady state after titration to and maintenance of effective pain relief.
Across the dose range administered to patients, there was no apparent correlation between hydromorphone ER dose and AUEC for pain intensity. Because the inclusion of conversion, titration, and maintenance phases allowed patients to reach an individualized effective dose of hydromorphone ER at steady state before any pharmacodynamic assessments were made, this result was anticipated.Citation28 Indeed, dose–response relationships may be more likely when fixed-dose designs are employed. Nevertheless, these findings underscore current recommendations for individualized starting doses and titration of opioids for patients with chronic pain.Citation6
Rescue medication to manage breakthrough pain was used by 76.5% of patients. This was not unexpected given the incidence of breakthrough pain reported by other investigators.Citation29 Among patients in the present study who required rescue medication, there was no apparent correlation between hydromorphone ER dose and morphine dose used for the treatment of breakthrough pain, although the study was not specifically designed to detect such a relationship.
Hydromorphone ER was generally well tolerated, and concomitant medications, including antidepressants and cardiovascular or gastrointestinal/gastroesophageal medications, did not appear to affect the tolerability profile. This is particularly important because patients with chronic pain often receive treatment with several medications to effectively manage their overall health.Citation30 A total of 16 patients (72.7%) reported an AE during the study, all of which were considered mild or moderate in severity. The most common AEs were constipation (31.8%), headache (22.7%), and vomiting (13.6%). Reported AEs were consistent with those expected for patients receiving potent opioid analgesics.Citation31 No serious AEs, unexpected AEs, or deaths occurred during the study.
This study had a number of limitations. Regarding the pharmacokinetic findings, the small number of patients receiving higher doses (24 mg and 48 mg) and the large proportion of female and white patients may limit the ability to generalize these findings to patients taking higher doses of hydromorphone ER and to other demographic segments of the population. The small number of patients and between-subjects design also affected the investigators’ ability to conduct formal analyses of dose proportionality. Regarding the pharmacodynamic results, although an increase in pain relief was observed, the lack of a placebo control did not allow for the assessment of potential improvements in patients not receiving active treatment. Additionally, this study evaluated hydromorphone ER for a relatively short period. However, previous studies have demonstrated the efficacy of hydromorphone ER for up to 1 year.Citation25,Citation32,Citation33
These findings replicate and extend the steady-state pharmacokinetic profile data of hydromorphone ER, previously characterized in healthy volunteers, to include a population of patients with chronic pain. Consistent and sustained plasma concentrations were reported in this patient population with chronic pain taking a variety of peripherally and centrally acting concomitant medications. Hydromorphone ER was generally well tolerated, and AEs were consistent with those expected for a potent opioid analgesic. Taken together, these data indicate that hydromorphone ER can be safely administered to chronic pain patients taking multiple concomitant medications.
Acknowledgments
This study was supported by the Knoll Pharmaceutical Company (now part of Abbott Laboratories). Technical editorial and medical writing support for the development of this manuscript was provided by Amanda McGeary, MA, of Synchrony Medical, West Chester, PA. Funding for this support was provided by Mallinckrodt, a Covidien company, Hazelwood, MO.
Disclosure
The results of this study were presented at the American Pain Society 30th Annual Scientific Meeting, May 19–21, 2011, in Austin, TX, and at PAINWeek 2011, September 7–10, in Las Vegas, NV. Dr Vandenbossche discloses that he is an employee of Janssen Research and Development and a stockholder in Johnson and Johnson. Dr Richarz discloses that she is an employee of Janssen Global Services, LCC, and a stockholder in Johnson and Johnson. Dr Richards discloses that he is an employee of Johnson and Johnson Pharmaceutical Research and Development and a stockholder in Johnson and Johnson. The authors declare no other conflicts of interest in this work.
References
- TurkDCOkifujiAPain terms and taxonomies of painFishmanSMBallantyneJCRathmellJPBonica’s Management of Pain4th edBaltimore, MDLippincott Williams & Wilkins20101323
- US Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Health StatisticsHealth, United States, 2006: with Chartbook on Trends in the Health of AmericansDHHS Publication No 2006-1232Hyattsville, MDNational Center for Health Statistics2006 Available from: http://www.cdc.gov/nchs/data/hus/hus06.pdfAccessed June 27, 2011
- US Census BureauPreliminary Annual Estimates of the Resident Population for the United States, Regions, States, and Puerto Rico: April 1, 2000 to July 1, 2010 (NSTPEST2010-01)2002 estimate Available from: http://www.census.gov/popest/research/eval-estimates/eval-es+2010.htmlLast accessed April 4, 2012
- McCarbergBHNicholsonBDToddKHPalmerTPenlesLThe impact of pain on quality of life and the unmet needs of pain management: results from pain sufferers and physicians participating in an Internet surveyAm J Ther200815431232018645331
- TurkDCWilsonHDCahanaATreatment of chronic non-cancer painLancet201137797842226223521704872
- ChouRFanciulloGJFinePGAmerican Pain Society-American Academy of Pain Medicine Opioids Guidelines PanelClinical guidelines for the use of chronic opioid therapy in chronic noncancer painJ Pain200910211313019187889
- ArgoffCESilversheinDIA comparison of long- and short-acting opioids for the treatment of chronic noncancer pain: tailoring therapy to meet patient needsMayo Clin Proc200984760261219567714
- HerreroJFLairdJMLópez-GarcíaJAWind-up of spinal cord neurones and pain sensation: much ado about something?Prog Neurobiol200061216920310704997
- FinePGMahajanGMcPhersonMLLong-acting opioids and short-acting opioids: appropriate use in chronic pain managementPain Med200910Suppl 2S79S8819691687
- QuigleyCWiffenPA systematic review of hydromorphone in acute and chronic painJ Pain Symptom Manage200325216917812590032
- QuigleyCHydromorphone for acute and chronic painCochrane Database Syst Rev20021CD00344711869661
- KnotkovaHFinePGPortenoyRKOpioid rotation: the science and the limitations of the equianalgesic dose tableJ Pain Symptom Manage200938342643919735903
- LawlorPTurnerKHansonJBrueraEDose ratio between morphine and hydromorphone in patients with cancer pain: a retrospective studyPain1997721–279859272790
- BrueraEPereiraJWatanabeSBelzileMKuehnNHansonJOpioid rotation in patients with cancer pain. A retrospective comparison of dose ratios between methadone, hydromorphone, and morphineCancer19967848528578756381
- MercadanteSCaraceniAConversion ratios for opioid switching in the treatment of cancer pain: a systematic reviewPalliat Med201125550451521708857
- MahlerDLForrestWHJrRelative analgesic potencies of morphine and hydromorphone in postoperative painAnesthesiology197542560260748347
- Exalgo (hydromorphone HCl) extended-release tablets CII [package insert]Hazelwood, MOMallinckrodt2010
- DroverDRAngstMSValleMInput characteristics and bioavailability after administration of immediate and a new extended-release formulation of hydromorphone in healthy volunteersAnesthesiology200297482783612357147
- SathyanGXuEThipphawongJGuptaSKPharmacokinetic investigation of dose proportionality with a 24-hour controlled-release formulation of hydromorphoneBMC Clin Pharmacol20077317270058
- MooreKTSt-FleurDMarriccoNCSteady-state pharmacokinetics of extended-release hydromorphone (OROS hydromorphone): a randomized study in healthy volunteersJ Opioid Manag20106535135821046932
- CollinsSLFauraCCMooreRAMcQuayHJPeak plasma concentrations after oral morphine: a systematic reviewPain Symptom Manage1998166388402
- TanGJensenMPThornbyJLShantiBFValidation of the Brief Pain Inventory for chronic nonmalignant painJ Pain20045213313715042521
- NCI Thesaurus, Hoechst Adverse Reaction Terminology SystemThe National Center for Biomedical Ontology BioPortal website. http://bioportal.bioontology.org/ontologies/46317/?p-terms&conceptid=Hoescht_Adverse_Reaction_Terminology_SystemAdverse Reaction Terminology SystemAccessed October 5, 2012
- GuptaSSathyanGProviding constant analgesia with OROS® hydromorphoneJ Pain Symptom Manage200733Suppl 2S19S24
- HannaMTucaAThipphawongJAn open-label, 1-year extension study of the long-term safety and efficacy of once-daily OROS(R) hydromorphone in patients with chronic cancer painBMC Palliat Care200981419754935
- TurgeonJGröningRSathyanGThipphawongJRicharzUThe pharmacokinetics of a long-acting OROS hydromorphone formulationExpert Opin Drug Deliv20107113714419961358
- AngstMSDroverDRLötschJPharmacodynamics of orally administered sustained-release hydromorphone in humansAnesthesiology2001941637311135723
- TingNDose Finding in Drug DevelopmentNew York, NYSpringer2006
- PortenoyRKBennettDSRauckRPrevalence and characteristics of breakthrough pain in opioid-treated patients with chronic noncancer painJ Pain20067858359116885015
- Parsells KellyJCookSFKaufmanDWAndersonTRosenbergLMitchellAAPrevalence and characteristics of opioid use in the US adult populationPain2008138350751318342447
- KalsoEEdwardsJEMooreRAMcQuayHJOpioids in chronic non-cancer pain: systematic review of efficacy and safetyPain2004112337238015561393
- RicharzUWaechterSSabatowskiRSzczepanskiLBinsfeldHSustained Safety and Efficacy of Once-Daily Hydromorphone Extended-Release (OROS(®) hydromorphone ER) Compared with Twice-Daily Oxycodone Controlled-Release Over 52 Weeks in Patients with Moderate to Severe Chronic Noncancer PainPain Pract2012 Epub Apr 18
- WallaceMMoulinDERauckRLLong-term safety, tolerability, and efficacy of OROS hydromorphone in patients with chronic painJ Opioid Manag2009529710519507806