Abstract
Background
Celecoxib exerted analgesic effects (hypoalgesia) reversed by opioid receptor antagonists in a model of inflammatory pain. To analyze this celecoxib-induced hypoalgesia further, we assessed the effects of several disruptors of cytoskeletal components in our model of inflammation.
Methods
Hyperalgesia to mechanical stimuli was induced in rat hind paws by intraplantar injection of carrageenan and measured using the Randall-Selitto method over the next 8 hours. The effects of systemic pretreatment with celecoxib and a range of cytoskeletal disruptors (colchicine, nocodazole, cytochalasin B, latrunculin B, acrylamide, each given by intraplantar injection) on carrageenan-induced hyperalgesia were similarly investigated. Morphine and other selective cyclo-oxygenase 1 (SC-560), cyclo-oxygenase 2 (SC-236), and nonselective cyclo-oxygenase (indomethacin) inhibitors were also tested under similar conditions.
Results
None of the cytoskeletal disruptors affected the peak intensity of carrageenan-induced hyperalgesia, and its duration was increased only by nocodazole and colchicine. Pretreatment with celecoxib 30 minutes before carrageenan reversed the hyperalgesia and raised the nociceptive threshold (hypoalgesia). All analgesic effects of celecoxib were blocked by nocodazole, colchicine, cytochalasin B, and latrunculin B. Pretreatment with morphine also induced hypoalgesia in carrageenan-inflamed paws, an effect reversed by colchicine and cytochalasin B. However, the analgesic effects of indomethacin were not reversed by disruption of actin filaments with cytochalasin B or latrunculin B.
Conclusion
These data strengthen the correlation between cytoskeletal structures and the processes of pain and analgesia.
Introduction
Celecoxib is classified as a nonsteroidal anti-inflammatory drug (NSAID), and its analgesic and anti-inflammatory actions have been attributed to its inhibition of prostaglandin biosynthesis via cyclo-oxygenase (COX), in common with other NSAIDs, such as indomethacin.Citation1,Citation2 We have unexpectedly found that analgesia induced by celecoxib in a model of inflammatory pain was, in contrast with that induced by indomethacin, reversed by opioid receptor antagonists and was not observed in animals tolerant to morphine.Citation3–Citation5 We have inferred from these results that the analgesia induced by celecoxib is mediated by endogenous opioids, presumably indirectly, involving release from opioidergic nerves because celecoxib is not a direct agonist of opioid receptors.Citation6 However, the mechanistic details of such an indirect action are not known, and in our continuing analysis of celecoxib-induced analgesia, we have investigated the effects of celecoxib on cellular mechanisms known to be involved in antinociception which are not primarily linked to prostaglandin biosynthesis or COX inhibition.
One such mechanism is the rearrangement of intracellular structures, collectively called the cytoskeleton, which appears to be involved in the cellular signaling associated with inflammatory pain.Citation7–Citation9 Therefore, we assessed the effects of disruptors of cytoskeletal components and their functions (referred to as cytoskeletal disruptors for brevity) on the analgesia induced by celecoxib in our model of inflammatory pain in rat paws. We studied three types of cytoskeletal disruptors targeted to particular components of the cytoskeleton, ie, nocodazole, and colchicine for microtubules,Citation10,Citation11 cytochalasin B and latrunculin B for actin filaments,Citation12–Citation14 and acrylamide for intermediate filaments,Citation15,Citation16 each given locally into the paw. In this model of hyperalgesia following injection of carrageenan in rat paws,Citation17,Citation18 we also used, for comparison, other known analgesic compounds, ie, morphine and NSAIDs, including indomethacin and the SC-560Citation19 and SC-236Citation2 compounds (selective inhibitors of COX-1 and COX-2, respectively).
Our results showed that some, but not all, of the cytoskeletal disruptors interacted with our model, affecting the hyperalgesia induced by carrageenan and the analgesia induced by celecoxib and morphine but not that induced by indomethacin or SC-560. Overall, our results add to the growing evidence for a causal and critical involvement of the cytoskeleton in the processes of nociception and analgesia in the periphery. Furthermore, the similarity of responses to the cytoskeletal disruptors shown by celecoxib and morphine give further support to our proposal that, in this model, the analgesic activity of celecoxib, unlike that of indomethacin, is mediated by endogenous opioids. Some preliminary data from this study have already appeared in abstract form.Citation20
Materials and methods
Animals
All animal procedures conformed to the guidelines of the International Association for the Study of PainCitation21 and were approved by the Federal University of Minas Gerais ethics committee for animal experimentation (protocol 180/07). Male Holtzman rats weighing 160–190 g were randomly divided into groups of five animals and housed under conditions of a 12-hour light/dark cycle and a temperature of 24°C ± 2°C. The animals were allowed free access to food and water. Three days before the experiments, the rats were exposed to the experimental room and apparatus and to handling by the investigator.
Inflammatory pain model
A standard hyperalgesic effect was produced by intraplantar injection of λ-carrageenanCitation3 250 μg in 0.1 mL of sterile saline into one hind paw at time zero. The contralateral paws were injected with the same interplantar volume of saline.
Measurement of mechanical nociceptive response
Assays to determine the nociceptive thresholds have been described in detail elsewhere.Citation3,Citation4,Citation22 Essentially, we measured the force (in g) applied to the plantar paw surface necessary to trigger hind paw withdrawal using a Basile algesimeter (Ugo Basile SRL, Comerio VA, Italy).Citation23 In order to avoid tissue damage, the maximum force applied was limited to 300 g. The hyperalgesic response to carrageenan is shown as a fall in nociceptive thresholds and the term “hypoalgesia” is used to describe the condition when the nociceptive threshold was raised above the basal level, ie, the level at time zero before any treatment of the animal. “Antihyperalgesia” in treated animals is defined as any reversal of hyperalgesia, up to the basal nociceptive threshold. The nociceptive response was measured before, then 30 minutes and one, 2, 3, 4, 6 and 8 hours after carrageenan or saline injections. Results for each paw are presented separately when the nociceptive threshold was affected in both paws, as after morphine (see below). For all experiments showing unilateral effects, the difference (λ) between nociceptive thresholds in the right and left paw is presented. Data are presented as time courses.
Hind paw edema measurements
The volume (in mL) of the hind paws for both the vehicle and treated animals was measured with a hydroplethysmometer (Ugo Basile 1750, Italy) at the same time points used for nociceptive threshold measurements, ie, time zero and one, 2, 3, 4, and 6 hours after stimulus injection. Results are presented as the difference (λ, mean ± standard error of the mean) in volume between the test (right) paw and the control (left) paw for each group of animals, at the time shown.
Drug doses, route of administration, and schedules
Cytoskeletal disruptors
All compounds were injected in a 0.1 mL volume of saline via the intraplantar route. We used colchicine (0.8 μg and 8 μg [2.03 nmoles and 20.3 nmoles]) and nocodazole (1 μg and 10 μg [3.32 nmoles and 33.2 nmoles]) as disruptors of microtubules; latrunculin B (0.5–5 μg [0.012–1.26 nmoles]) and cytochalasin B (1 ng–1 μg [0.002–2.09 nmoles]) as disruptors of actin filaments and phalloidin (0.1–10 μg [0.13–12.7 nmoles]) as a stabilizer of actin filaments; and acrylamide (0.1–10 μg [1.4–141 nmoles]) as a disruptor of intermediate filaments. All these compounds were stored as stock solutions in dimethyl sulfoxide at −15°C. On the day of the experiment, they were diluted with sterile saline; the final maximum concentration of dimethyl sulfoxide was 1%, and this concentration did not affect the nociceptive thresholds.
COX inhibitors
All compounds were systemically injected s.c. in a volume of 0.1 mL per 100 g animal. We used the standard nonselective inhibitor, indomethacin (4 mg/kg), a selective COX-1 inhibitor, SC-560 (5 mg/kg), and two selective COX-2 inhibitors, celecoxib (12 mg/kg) and SC-236 (12 mg/kg). Indomethacin, SC-560, and SC-236 were dissolved in a mixture of 2% ethanol, 8% Tween 80, and 90% saline, and celecoxib was administered in saline.Citation3
Opioid drugs
Morphine 2 mg/kg was dissolved in saline and injected systemically at a volume of 0.1 mL/100 g animal.
Drug schedules
Cytoskeletal disruptors were injected into the right hind paw 30 minutes before systemic treatment with COX inhibitors or the opioid agonist, and the same right hind paw received intraplantar carrageenan 30 minutes thereafter. Contralateral paws were injected with the same volume of the corresponding vehicle.
Drug sources
The selective COX 1 and 2 inhibitors, SC-560 and SC-236, were purchased from Cayman Chemical Company (Ann Arbor, MI), celecoxib (Celebra™) from Pfizer Pharmaceuticals (Caguas, PR), and morphine hydrochloride from Merck AG, (Darmstadt, Germany). All cytoskeletal disruptors were purchased from Sigma-Aldrich (St Louis, MO).
Statistical analysis
Data are presented as the mean ± standard error of the mean and analyzed by one-way analysis of variance. Statistical comparisons of means were further examined by Bonferroni’s test. Statistical significance was accepted when the difference between means was less than 0.05 (P < 0.05).
Results
Hyperalgesia and edema induced by intraplantar carrageenan injection
A standard dose of carrageenan (250 μg per paw) was used in all the experiments, based on our earlier results.Citation3,Citation4 This dose induced a characteristic fall in the nociceptive threshold, compared with the contralateral, saline-injected paws, with maximal hyperalgesia reached 2–3 hours after carrageenan injection, returning to normal basal values between 6 and 8 hours (). Carrageenan also induced edema, assayed as increased paw volume (), over the same time course. This aspect of the inflammatory response peaked at 3 hours after carrageenan injection and was still detectable at 6 hours. Paw volumes of the left noninflamed paw which received only saline did not change over the time course of the experiments (data not shown).
Figure 1 Colchicine and nocodazole potentiate carrageenan-induced hyperalgesia in rat paws.
Abbreviations: Veh, vehicle; CCC, colchicine; CG, carrageenan; NDZ, nocodazole.
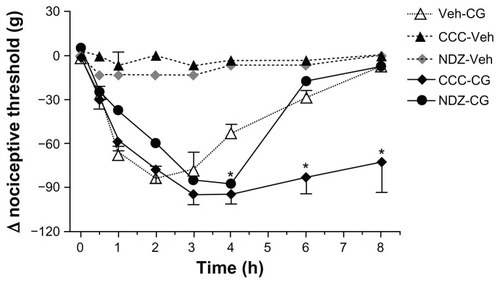
Table 1 Effects of cytoskeletal disruptors, given locally, on carrageenan-induced edema in rat paws
Local injection of cytoskeleton disruptors and inflammatory response to carrageenan
We assessed first the effects of local intraplantar injection of cytoskeletal disruptors on basal nociceptive threshold and on the hyperalgesia induced by carrageenan. None of the compounds used affected basal thresholds, ie, those measured in paws injected with saline, assayed over 8 hours or the absolute values at time zero in groups of treated animals (data not shown). However, carrageenan-induced hyperalgesia was modified by pretreatment with cytoskeletal disruptors, but only by those affecting microtubule assembly, ie, nocodazole and colchicine (). As the time course for these two compounds shows, the peak intensity of hyperalgesia in the early stages (up to 3 hours after carrageenan) was not changed but the duration of hyperalgesia was extended, most clearly by colchicine. The corresponding time courses for the other compounds showed no changes from the time course of carrageenan given alone (data not shown). The contralateral paws, injected with saline instead of carrageenan, did not show changes in nociceptive threshold after carrageenan or after any of the cytoskeletal disruptors (data not shown). The cytoskeletal disruptors produced a comparable profile of effects on the edema induced by carrageenan (). Only nocodazole or colchicine affected this response and both compounds potentiated or prolonged the increased volume of the inflamed paw. No changes were induced in the volumes of the noninflamed paw by any of the cytoskeletal disruptors tested (data not shown).
Effect of cytoskeletal disruptors on analgesic effects of celecoxib
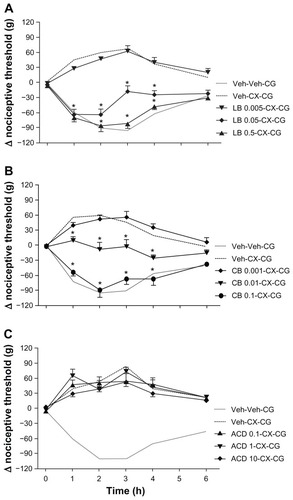
We next tested the effects of the cytoskeletal disruptors on the characteristic hypoalgesia induced by celecoxib. The two disruptors of microtubule assembly, nocodazole and colchicine, dose-dependently prevented the hypoalgesic response following systemic administration of celecoxib to rats injected with carrageenan (). Both compounds at the higher doses abolished all the analgesic effects of celecoxib, and the nociceptive thresholds after celecoxib and colchicine were almost identical to those seen in the absence of celecoxib (see ). The disruptors of actin filaments, ie, latrunculin B and cytochalasin B, also achieved dose-dependent reversal of the analgesic effects of celecoxib (). It is worth noting that these two compounds were, in molar terms, almost 10-fold more potent than nocodazole and colchicine in reversing celecoxib-induced antinociception. Pretreatment with acrylamide, a disruptor of intermediary filaments, over a range of doses did not affect the hypoalgesic effect of celecoxib (). Because our experiments showed that cytochalasin B was a potent inhibitor of the hypoalgesia induced by celecoxib and by itself did not modify the hyperalgesia induced by carrageenan in rat paws, we used cytochalasin B as a standard cytoskeleton disruptor in our subsequent studies.
Figure 2 Reversal of celecoxib-induced hypoalgesia by nocodazole or colchicine. Celecoxib 12 mg/kg administered systemically 30 minutes before carrageenan prevented development of hyperalgesia after carrageenan and induced hypoalgesia (λ nociceptive threshold values greater than zero). In (A), pretreatment with intraplantar nocodazole 1 μg or 10 μg administered 60 minutes before carrageenan dose-dependently reversed the analgesic effects of celecoxib. Data are shown as the mean ± standard error of the mean for five rats in each treatment group. *P < 0.05, significant effect of nocodazole. Similarly in (B), intraplantar colchicine 0.8 μg or 8 μg administered 60 minutes before carrageenan reversed the effects of celecoxib. Data are shown as the mean ± standard error of the mean for five rats in each treatment group. *P < 0.05, significant effects of colchicine.
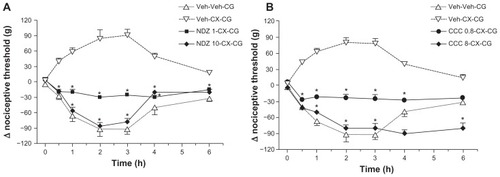
Figure 3 Effects of latrunculin B, cytochalasin B, and acrylamide on celecoxib-induced hypoalgesia. Although intraplantar latrunculin B 0.005–0.5 μg administered 60 minutes before carrageenan did not modify the hyperalgesia induced by carrageenan (data not shown), in (A), it dose-dependently reversed the hypoalgesic effects of celecoxib 12 mg/kg administered systemically 30 minutes before carrageenan. Similarly in (B), intraplantar cytochalasin B 0.001–1 μg administered 60 minutes before carrageenan, which did not affect carrageenan-induced hyperalgesia (data not shown), dose-dependently reversed celecoxib-induced hypoalgesia. However, intraplantar acrylamide 0.1–10 μg administered 60 minutes before carrageenan neither affected carrageenan-induced hyperalgesia (data not shown) nor, as shown in (C), the corresponding celecoxib-induced hypoalgesia.
Abbreviations: ACD, acrylamide; CB, cytochalasin B; CCC, colchicine; CG, carrageenan; CX, celecoxib; LB, latrunculin B; Veh, vehicle.
Time dependence of celecoxib-induced reversal of hypoalgesia by cytochalasin B
In our first set of experiments with cytochalasin B, we used it as a pretreatment, ie, before either celecoxib or carrageenan. We now gave it after carrageenan, at times when the celecoxib-induced hypoalgesia was developing, at 30 or 60 minutes following carrageenan. As shown in , treatment at these later times was less effective in reversing hypoalgesia due to celecoxib. When cytochalasin B was given at 30 minutes after carrageenan, directly after the assessment of nociceptive threshold, hypoalgesia was already present and 30 minutes later (at one hour after carrageenan) the thresholds were close to those after carrageenan alone, and at all other times there was full reversal of celecoxib-induced hypoalgesia. Given at 60 minutes, at the peak of hypoalgesia, the same dose of cytochalasin B only partly reversed celecoxib-induced analgesia. In addition, at neither of these times was cytochalasin B, given to the contralateral paw, able to modify the nociceptive response to carrageenan or the hypoalgesia induced by celecoxib (data not shown). Phalloidin, a stabilizer of actin filaments, has been shown to block the effects of cytochalasin D in epinephrine-induced paw hyperalgesia.Citation16 In our model, similar doses of phalloidin did not affect basal nociceptive thresholds for carrageenan-induced hyperalgesia (data not shown) or celecoxib-induced hypoalgesia (), but it was able to prevent cytochalasin B from reversing celecoxib-induced analgesia (). In these experiments, intraplantar cytochalasin B was again given 30 minutes before carrageenan, so that the number of pretreatment injections into the paw was not increased.
Figure 4 Reversal by cytochalasin B of celecoxib-induced hypoalgesia is dependent on time of treatment.
Abbreviations: CG, carrageenan; CX, celecoxib; LB, latrunculin B; Veh, vehicle; CB, cytochalasin B.
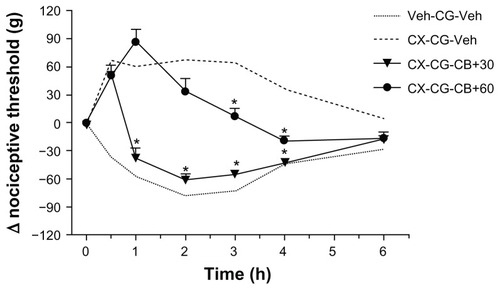
Figure 5 Effects of phalloidin on celecoxib-induced hypoalgesia and interactions with cytochalasin B. In (A), a range of intraplantar doses of phalloidin (0.1–10 μg) administered 60 minutes before carrageenan did not modify the hypoalgesia induced by celecoxib 12 mg/kg administered systemically 30 minutes before carrageenan. The data are shown as the mean ± standard error of the mean for five rats in each treatment group. In (B), intraplantar cytochalasin B 1 μg administered 30 minutes after carrageenan (see arrow on time axis) reversed all analgesic effects of celecoxib from 1–4 hours. Pretreatment with intraplantar phalloidin 1 μg administered 60 minutes before carrageenan blocked this reversal by cytochalasin B. The data are shown as the mean ± standard error of the mean for five rats in each treatment group.
Note: *P < 0.05, significant effect of phalloidin.
Effect of cytochalasin B on antinociceptive response induced by other nonselective and selective COX inhibitors
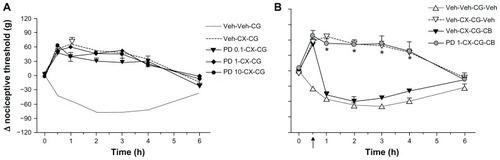
To assess the relevance of COX inhibition to the effects of cytochalasin B on celecoxib-induced hypoalgesia, we tested its effects on the analgesia known to be exerted in our model by a variety of COX inhibitors. As shown in , pretreatment with a local intraplantar injection of cytochalasin B did not affect the antinociceptive effects exerted by systemic treatment with doses of indomethacin (4 mg/kg), or the selective inhibitor of COX-1, SC-560 (5 mg/kg). However, hypoalgesia induced by SC-236, another selective inhibitor of COX-2, which is as effective as celecoxib in our model,Citation3–Citation5 was reversed by cytochalasin B.
Figure 6 Effects of pretreatment with cytochalasin B on the analgesic effects of other COX inhibitors in carrageenan-induced hyperalgesia.
Abbreviations: AUC, area under the concentration-time curve; CB, cytochalasin B; CG, carrageenan; COX, cyclo-oxygenase; Indo, indomethacin; Veh, vehicle.
Interactions between cytoskeletal disruptors and opioid system in carrageenan-induced hyperalgesia
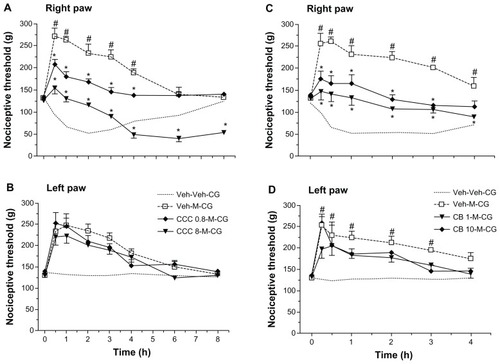
Morphine given as a pretreatment in our model of carrageenan-induced hyperalgesia has a bilateral hypoalgesic effect in rats following intracerebroventricular administration.Citation5,Citation24 To assess the effects of cytoskeletal disruption on this hypoalgesia when induced by morphine, we used cytochalasin B, colchicine, and acrylamide. Systemic morphine 2 mg/kg induced hypoalgesia in both carrageenan-injected (right, inflamed) rat paws () and saline-injected (left, noninflamed) rat paws (). Colchicine dose-dependently inhibited morphine-induced hypoalgesia and, at the higher dose and at the later times (4 hours onward), produced the now characteristic prolongation of hyperalgesia, up to 8 hours after carrageenan (). Cytochalasin B, over the same dose range that totally reversed celecoxib-induced hypoalgesia was less effective against morphine-induced hypoalgesia, although there was still a very marked decrease in hypoalgesia without complete reversal of all analgesic effects (). Note that neither of these cytoskeletal disruptors, given only to the inflamed paw, affected the response to morphine in the contralateral paw (). The disruptor of intermediate filaments, acrylamide, given at 1 μg or 10 μg to the inflamed paw, did not affect morphine-induced hypoalgesia in either the right or left paws (data not shown).
Figure 7 Modulation of morphine-induced hypoalgesia after carrageenan by colchicine or cytochalasin B. In (A), morphine (M) 2 mg/kg administered systemically 30 minutes before carrageenan (CG) was clearly analgesic, changing the hyperalgesia after carrageenan to hypoalgesia. Pretreatment with two doses of intraplantar colchicine (CCC) 0.8 μg or 8 μg administered at 60 minutes before carrageenan decreased the hypoalgesia but retained the antihyperalgesic effects of morphine. In (B), data from the left paw (without carrageenan) show that hypoalgesia after morphine was not changed by colchicine. Data are shown as the mean ± standard error of the mean for five rats in each treatment group. Similarly, in (C and D), intraplantar cytochalasin B (CB) 1 μg or 10 μg administered 60 minutes before carrageenan partly reversed morphine-induced analgesia to antihyperalgesia in the inflamed right paw, without modifying the effects of morphine in the left noninflamed paw. Data are shown as the mean ± standard error of the mean for five rats in each treatment group.
Abbreviations: CB, cytochalasin B; CCC, colchicine; CG, carrageenan; Veh, vehicle.
Discussion
Our experiments have shown that disruption of the cytoskeleton reversed the analgesia induced by two selective inhibitors of COX-2, ie, celecoxib and SC-236. Such reversal was selective in that not all the cytoskeletal disruptors were equally effective and that the analgesic effects of the nonselective COX inhibitor, indomethacin, and of SC-560, were not affected by any of the cytoskeletal disruptors. However, cytoskeletal disruption also reversed morphine-induced analgesia, strengthening the causal relationship between cytoskeletal structure and the processes involved in nociception and endogenous antinociceptive systems.
Our first experiments evaluating the effects of different cytoskeletal disruptors on the nociceptive thresholds to mechanical stimuli under basal conditions and after inflammation with carrageenan highlighted clear differences between their effects in inflamed paws, although none of them modulated nociceptive thresholds in saline-injected control rat paws. Such control responses were observed either in the left (saline-injected) paw of rats receiving carrageenan in the right paw or in rats receiving saline in only one hind paw. From these results, we deduced that the inflammation was localized, ie, restricted to the injected paw without signs of a systemic response to carrageenan. This lack of effect in control paws would also imply that the cytoskeletal disruptors and, hence, the need for specific cytoskeletal rearrangement, were not critical for the normal nociceptive response to mechanical stimulation in normal noninflamed paws. Our results would agree with those of Dina et al,Citation16 who reported a similar lack of modulation of basal nociception by these cytoskeletal disruptors. However, in paws treated with carrageenan, disruptors of microtubules (nocodazole and colchicine) were very clear potentiators of carrageenan-induced hyperalgesia, whereas the others (cytochalasin B, latrunculin B, and acrylamide) did not modify the hyperalgesic effects of carrageenan. Because we measured the time course of hyperalgesia over 6 hours, rather than making a single fixed time assay, we were able to show that the potentiation of carrageenan hyperalgesia was not expressed as a change in intensity of the hyperalgesia, but as an extension of its duration. Our model is capable of showing an increased intensity of hyperalgesia, for instance, to higher doses of carrageenan than those used here.Citation3 This increased duration of hyperalgesia might suggest that the major effect of the disrupted microtubules was to influence the resolution of, or recovery from, the hyperalgesic state, rather than its initiation or generation of mediators of hyperalgesia. Such modification of carrageenan-induced inflammation is supported by the effects of the cytoskeletal disruptors on paw edema, where only nocodazole and in particular colchicine prolonged the edema (increased paw volume) in the carrageenan-injected paw. Given that pain and edema are two of the cardinal signs of inflammation, it would appear that disruption of microtubules selectively inhibited resolution of the inflammatory process. This conclusion is in marked contrast with the long established clinical use of systemic colchicine as a potent anti-inflammatory agent.Citation11,Citation25,Citation26 This difference is most likely to be related to the different modes of administration; we gave colchicine locally to the paw, whereas in clinical use, colchicine is given orally. Systemic colchicine would clearly have as its first targets circulating leukocytes and endothelial cells, two well recognized sites of the anti-inflammatory actions of colchicine.Citation11 Local intraplantar colchicine would access different types of cells and, in particular, relatively few leukocytes and endothelial cells. Interestingly, another potential proinflammatory action of colchicine is its inhibition of steroid biosynthesis,Citation27 but such action is probably not relevant over the short experimental period (6 hours) of our model. However, it would be of interest to assess the effects of systemic colchicine on nociception and edema in our model.
Our results with nocodazole and colchicine also contrast with those of Dina et al,Citation16 who found these two disruptors to be antinociceptive agents, reducing the hyperalgesia induced by epinephrine. However, these differences may be the result of the different algesic stimuli used (epinephrine, prostaglandin E2, estrogen), either alone or concomitantly, and the extent of the accompanying modulation of inflammation, all of which may involve different intracellular signaling pathways. For instance, peripheral nociception was differentially modulated when the paw was injected with carrageenan or prostaglandin E2.Citation28
We then used a range of known analgesic agents in our model and all clearly decreased the hyperalgesia, with some analgesic agents (celecoxib, SC-236, morphine) inducing a hypoalgesic effect. However, the cytoskeletal disruptors were not all equally effective in modulating these antinociceptive effects. Thus, the disruptors of microtubules or actin were more effective than acrylamide, which did not modify the antinociceptive effects of any of the analgesic agents used here.
Disruption of actin filaments by cytochalasin B or latrunculin B, although ineffective against carrageenan-induced hyperalgesia, proved to be highly effective in reversing the analgesic effects of celecoxib and SC-236. Moreover, phalloidin, an actin filament stabilizer,Citation12 prevented this effect of cytochalasin B, although phalloidin by itself did not affect celecoxib-induced hypoalgesia or the nociceptive response to carrageenan, implying that changes in actin filaments were induced particularly during the hypoalgesic response to celecoxib. A similar “antagonism” between phalloidin and cytochalasin D, another actin disruptor, has been reported in epinephrine-induced hyperalgesia.Citation16 The reversal of celecoxib-induced hypoalgesia by the two microtubule disruptors (nocodazole and colchicine) is less easily interpreted, because these compounds potentiated carrageenan-induced inflammation, ie, they prolonged the hyperalgesia and edema in the absence of any analgesic agent. Therefore, our results for nocodazole and colchicine were not immediately helpful in elucidating the mechanism(s) underlying the actions of celecoxib in our model. Nevertheless, low doses of cytochalasin B prevented celecoxib-induced hypoalgesia, an action that was completely blocked by phalloidin. This finding, taken together with the lack of effect for the intermediate filament disruptor, acrylamide, on celecoxib-induced hypoalgesia would suggest that actin filaments are crucially involved in the expression of celecoxib-induced hypoalgesia.
Another important outcome of these experiments was the clear division between the two coxibs (celecoxib and SC-236) and the other NSAIDs (indomethacin and SC-560), in terms of their response to cytochalasin B. Because indomethacin and SC-560 are inhibitors of COX,Citation19,Citation29 their analgesic activities have been attributed to their ability to block prostaglandin biosynthesis.Citation1 Thus, the resistance of analgesic effects to modification by cytochalasin B observed here would be compatible with the resistance of prostaglandin E2-induced paw hyperalgesia to cytochalasin D and other actin filament disruptors.Citation16 If, as proposed by Dina et al,Citation16 signaling pathways via protein kinase A are involved in prostaglandin E2-induced hyperalgesia and these are not affected by disruption of actin filaments, then equal resistance would be expected for those analgesics with an action based on lowering prostaglandin production. This division between the coxibs and the other NSAIDs would support the possibility of a nonprostaglandin-based mechanism underlying the celecoxib-induced hypoalgesia in this model, as we have consistently found.Citation3–Citation5,Citation30
The profile of effects of the cytoskeletal disruptors on morphine-induced analgesia in our model matched that obtained with celecoxib, insofar as disruptors of actin and microtubules reversed the effects of morphine, whereas acrylamide was ineffective. Although there is evidence of changes in cytoskeletal proteins after chronic morphine treatment in the context of correlating protein modifications with addiction and/or withdrawal,Citation31–Citation33 links between acute agonist exposure and proteins have focused on those proteins more closely associated with receptor function, such as β-arrestin, G protein-coupled inwardly-rectifying potassium (GIRK) channels, and regulators of G-protein signaling.Citation34,Citation35 Thus, our results for the acute modulation of morphine-induced analgesia by cytochalasin B may be the first such data from an in vivo model. Although the interactions of cytochalasin B with β-arrestin, GIRK, and G-protein signaling proteins have not been specifically studied, it is very likely that these cytoskeletal disruptors modulated the acute response to opioid receptor agonists by affecting actin and/or microtubular structures involved in the intracellular processing of such receptors through endosomes, because internalization and recycling of opioid receptors is crucial to their functionCitation36 and actin polymerization plays a critical role in endocytic function.Citation37,Citation38 Recently, rapid recycling of μ-opioid receptors in HEK293 cells and in neurons has been described, which involves a crucial interaction with actin and microtubules.Citation39 Disruption of these cytoskeletal components (with latrunculin and nocodazole) decreased recycling of internalized μ-receptors to the cell membrane, and could thus lead to a loss of response to opioid agonists. In our model, such events would lead to a loss of analgesia induced by morphine and endogenous opioids.
An important feature of the action of morphine in this model was that it affected nociceptive thresholds in the contralateral noninflamed paw as well as those in the inflamed paw, as found earlier.Citation5 It is worth noting that although morphine and celecoxib were both given systemically, celecoxib only affected thresholds in the inflamed paw,Citation3–Citation5,Citation40 and this difference does imply differences in the sites of action between the two compounds. In the present experiments, colchicine and cytochalasin B were relatively less effective against morphine than they were against celecoxib, with some antinociceptive effects persisting for the first few hours after morphine. Bearing in mind that these two disruptors were given locally into the paw, this difference could derive from a difference in action on peripheral versus central sites. Although both morphine and celecoxib are active peripherally and centrally,Citation5,Citation24,Citation41,Citation42 morphine is more likely to access the central nervous system rapidly and is a direct agonist at opioid receptors, whereas celecoxib does not readily cross the blood–brain barrierCitation5,Citation43,Citation44 and must act indirectly because it is not an agonist at opioid receptors.Citation6 These differences may result in a greater proportion of centrally mediated analgesia for morphine than for celecoxib, when either is given systemically. Only the local peripheral actions of the analgesics would be affected by cytochalasin B given via the intraplantar route, because our assay with cytochalasin B given to the noninflamed paw showed that not enough cytochalasin B circulated to the inflamed paw to modulate nociceptive responses. The less efficient reversal of the effect of morphine probably reflects the greater proportion of its analgesia being mediated via the central nervous system.
One clear limitation of our study is that although we considered the actions of the cytoskeletal disruptors to be restricted to the paw and thus acting peripherally, we do not know which cells in the paw are primarily affected. Given that cytochalasin B reversed morphine-induced analgesia, one site of action for the cytoskeletal disruptors could be cells bearing opioid receptors, most obviously, sensory neurones. Actions on such cells would also explain the reversal by cytochalasin B of celecoxib-induced hypoalgesia, because this hypoalgesia was also blocked by opioid receptor antagonists.Citation4,Citation5,Citation30,Citation40 Furthermore, there is cross-tolerance between celecoxib and morphine.Citation45 Overall, there is good evidence in our model for celecoxib-induced hypoalgesia being mediated by activation of opioid receptors. However, because celecoxib is not an agonist at opioid receptors, we have postulated an indirect activation of opioid receptors by the release of endogenous opioids.Citation5,Citation30 A relevant example of such release is the action of chemokines to release opioids from primary granules in polymorphonuclear leukocytes.Citation46 If, in our experimental system, celecoxib does release endogenous opioids from peripheral stores in leukocytesCitation46,Citation47 or keratinocytes,Citation48,Citation49 then the release process itself would provide another possible locus of action for the cytoskeletal disruptors. The involvement of actin in the release of two other peptides (oxytocin and vasopressin) from hypothalamic neuronesCitation50 and that of dopamine from PC12 cellsCitation51 has already been demonstrated.
There is very recent evidence for a further complication in the mechanism underlying celecoxib-induced hypoalgesia, because celecoxib given intracerebroventricularly also induced hypoalgesia in paws injected with carrageenan, and this central action of celecoxib was blocked by opioid receptor antagonists and by an antagonist of cannabinoid CB1 receptors.Citation30 These results would suggest that celecoxib may release endocannabinoids, which in turn, release endogenous opioids, as shown by Ibrahim et al.Citation49 If such a link can be demonstrated in the periphery, ie, after systemic administration of celecoxib, then there would be two levels of endogenous transmitter release and action at which cytochalasin B might act. Nevertheless, for either direct or indirect activation of opioid receptors, blockade of the intracellular processes following opioid receptor activation, by interfering with the cytoskeleton, would be sufficient to prevent analgesic effects.
Another limitation is that we have not shown directly that, in our model, the different cytoskeletal components were disrupted by the agents used. Also, several of the disruptors have other cellular effects, apart from any selective effects on the cytoskeleton. For instance, colchicine induces neuronal apoptosis and is antiproliferative,Citation11,Citation52 and nocodazole, colchicine, and cytochalasin B block glucose transport.Citation53 However, for colchicine and nocodazole, our finding that hyperalgesia and edema were prolonged rather than intensified or blocked suggests a more subtle effect than neuronal toxicity. For cytochalasin B, reversal by phalloidin would at least be compatible with effects on particular components of the cytoskeleton. Similarly, a general metabolic toxicity that could follow blockade of glucose uptake is also an unlikely explanation for our observations, given that glucose transporters are inhibited effectively by either nocodazole, colchicine or cytochalasin B,Citation53 whereas in our experiments, these cytoskeletal disruptors exerted clearly different effects. In future work, a direct assessment of the state of the microtubular system and actin in our experimental system should be undertaken.
In summary, our results imply that some, but not all, components of the cytoskeleton are involved in the mechanisms underlying the hyperalgesia and edema characteristic of a well established model of peripheral, localized inflammation. Furthermore, the analgesic effects of morphine and celecoxib, but not those of indomethacin, were also affected by disruption of the cytoskeleton. Overall, our results provide further evidence for the critical and causal involvement of the cytoskeleton in expression of inflammatory pain and of endogenous antinociceptive systems triggered by such stimuli. They also support our proposal that the mechanisms underlying the antinociceptive actions of celecoxib in this model involve the endogenous opioid system.
Acknowledgments
This work was supported by the Conselho Nacional de Pesquisa (National Council for Scientific and Technological Development [CNPq]), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Coordination for the Improvement of Higher Level Personnel [CAPES]), and the Fundação de Amparo à Pesquisa de Minas Gerais (The State of Minas Gerais Research Foundation [FAPEMIG]). The authors also acknowledge the excellent technical assistance of Webster Glayser Pimenta dos Reis.
Disclosure
The authors report no conflicts of interest in this work.
References
- VaneJRBakhleYSBottingRMCyclooxygenases 1 and 2Annu Rev Pharmacol Toxicol199838971209597150
- PenningTBTalleyJJBertenshawSRSynthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-y1] benzenesulfonamide (SC-58635, Celecoxib)J Med Chem199740347365
- FrancischiJNChavesCTLimaASBakhleYSA new anti-nociceptive pathway mediated by cyclooxygenase-2 in rat paw inflammation induced by carrageenanBr J Pharm2002135159
- FrançaDSFerreira-AlvesDLDuarteIDEndogenous opioids mediate the hypoalgesia induced by inhibitors of cyclooxygenase-2 in rat paws treated with carrageenanNeuropharmacology200651374316620880
- RezendeRMReisWGPDuarteIDLimaPPBakhleYSFrancischiJNThe analgesic actions of centrally administered celecoxib are mediated by endogenous opioidsPain20091429410019186002
- Gregori-PuigjanéEMestresJA ligand-based approach to mining the chemogenomic space of drugsComb Chem High Throughput Screen20081166967618795886
- BhaveGGereauRWGrowing pains: cytoskeleton as a critical regulator of pain plasticityNeuron20033957758312925270
- HuchoTLevineJDSignalling pathways in sensitization: towards a nociceptor cell biologyNeuron20075536537617678851
- EisenhutMWallaceHIon channels in inflammationPflugers Arch201146140142121279380
- SamsonFDonosoJABettingerIHWatsonDHimesRHNocodazole action on tubulin assembly, axonal ultrastructure and fast axoplasmic transportJ Pharmacol Exp Ther197920841141785702
- TerkeltaubRAColchicine update: 2008Semin Arthritis Rheum20083841141918973929
- CooperJAEffects of cytochalasin and phalloidin on actinJ Cell Biol1987105147314783312229
- SpectorIShochetNRBlasbergerDKashmanYLatrunculins – novel marine macrolides that disrupt microfilament organization and affect cell growth: I. Comparison with cytochalasin DCell Motil Cytoskeleton1989131271442776221
- WakatsukiTSchwabBThompsonNCElsonELEffects of cytochalasin D and latrunculin B on mechanical properties of cellsJ Cell Sci20011141025103611181185
- LoPachinRMRossJFLehningEJNerve terminals as the primary site of acrylamide action: a hypothesisNeurotoxicology200223435912164547
- DinaAOMcCarterGCCoupadeCLevineJDRole of the sensory neuron cytoskeleton in second messenger signalling for inflammatory painNeuron20033961362412925276
- Di RosaMBiological properties of carrageenanJ Pharm Pharmacol197224891024402944
- VinegarRTruaxJFSelphJLJohnstonPRVenableALMcKenzieKKPathway to carrageenan-induced inflammation in the hind limb of the ratFed Proc1987461181263100339
- SmithCJZhangYKoboldtCMPharmacological analysis of cyclooxygenase-1 in inflammationProc Natl Acad Sci U S A19989513313133189789085
- FrancischiJNPaiva-LimaPFrançaDSBakhleYSCrucial role of the cytoskeleton in hypoalgesia induced by celecoxib in rat inflammatory painVIIIth World Congress of InflammationCopenhagenInflammation Res200756389
- ZimmermannMEthical guidelines for investigations of experimental pain in conscious animalsPain1983161091106877845
- RezendeRMFrançaDSMenezesGBDos ReisWPGBakhleYSFrancischiJNDifferent mechanisms underlie the analgesic actions of paracetamol and dipyrone in a rat model of inflammatory painBr J Pharmacol20085376076818157167
- RandallLDSelittoJJA method for measurement of analgesic activity on inflamed tissuesArch Int Pharmacodyn Ther195711323324913509788
- FerreiraSHLorenzettiBBCorrêaFMACentral and peripheral antialgesic action of aspirin-like drugsEur J Pharmacol1978533948310771
- AhernMJReidCGordonTPMcCredieMBrooksPMJonesMDoes colchicine work? The results of the first controlled study in acute goutAust N Z J Med1987173013043314832
- DasSKRamakrishnanSMishraKA randomized controlled trial to evaluate the slow-acting symptom-modifying effects of colchicine in osteoarthritis of the knee: a preliminary reportArthritis Rheum20024728028412115158
- SewerMBLiDRegulation of steroid hormone biosynthesis by the cytoskeletonLipids2008431109111518726632
- AlvesDPMottaPGPaiva-LimaPInflammation mobilizes local resources to control hyperalgesia: the role of endogenous opioid peptidesPharmacology201289222822236644
- VaneJRInhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugsNat New Biol19712312322355284360
- RezendeRMLimaPPReisWGPEndogenous opioid and cannabinoid mechanisms are involved in the analgesic effects of celecoxib in the central nervous systemPharmacology20128912713622415159
- LoguinovAVAndersonLMCrosbyGJYukhananovRYGene expression following acute morphine administrationPhysiol Genomics2001616918111526201
- Marie-ClaireCCourtinCRoquesBPNobleFCytoskeletal genes regulation by chronic morphine treatment in rat striatumNeuropsychopharmacology2004292208221515199374
- ChristieMJCellular neuroadaptations to chronic opioids: tolerance, withdrawal and addictionBr J Pharmacol200815438439618414400
- González-RodriguesSHidalgoABaamondeAMenéndezLInvolvement of Gi/o proteins and GIRK channels in the potentiation of morphine-induced spinal analgesia in acutely inflamed miceNaunyn Schmiedebergs Arch Pharmacol2010381597119940980
- FanXLZhangJSZhangXQYueWMaLDifferential regulation of beta-arrestin 1 and beta-arrestin 2 gene expression in rat brain by morphineNeuroscience200311738338912614678
- NagiKPiñeyroGRegulation of opioid receptor signalling: implications for the development of analgesic toleranceMol Brain201142521663702
- PutthenveeduMALaufferBTemkinPSequence-dependent sorting of recycling proteins by actin-stabilized endosomal microdomainsCell201014376177321111236
- SkruznyMBrachTCiuffaRRybinaSWachsmuthMKaksonenMMolecular basis for coupling the plasma membrane to the actin cytoskeleton during clathrin mediated endocytosisProc Natl Acad Sci U S A2012109E2533E254222927393
- Roman-VendrellCYuYJYudowskiGAFast modulation of μ-opioid receptor (MOR) recycling is mediated by receptor agonistsJ Biol Chem2012287147821479122378794
- CorreaJDPaiva-LimaPRezendeRMPeripheral mu-, kappa- and delta-opioid receptors mediate the hypoalgesic effect of celecoxib in a rat model of thermal hyperalgesiaLife Sci20108695195620451533
- SofiaRDVassarHBKnoblochLCComparative analgesic activity of various naturally occurring cannabinoids in mice and ratsPsychopharmacologia1975402852951170585
- LabuzDMousaASSchäferMSteinCMachelskaHRelative contribution of peripheral versus central opioid receptors to antinociceptionBrain Res20071160303817599812
- BinghamSBeswickPJBountraCThe cyclooxygenase-2 inhibitor GW406381X [2-(4-ethoxyphenyl)-3-[4-(methylsulfonyl)phenyl]-pyrazolo[1,5-b]pyridazine] is effective in animal models of neuropathic pain and central sensitizationJ Pharmacol Exp Ther20053121161116915572651
- DemboGParkSBKharaschEDCentral nervous system concentrations of cyclooxygenase-2 inhibitors in humansAnesthesiology200510240941515681959
- RezendeRMPaiva-LimaPCamêloVMDos ReisWGPBakhleYSFrancischiJNCelecoxib induces tolerance in a model of peripheral inflammatory pain in ratsNeuropharmacology20105955155720691196
- RittnerHLLabuzDSchaeferMPain control by CXCR2 ligands through Ca2+-regulated release of opioid peptides from polymorphonuclear cellsFASEB J2006202627262917060402
- SteinCGramschCHerzAIntrinsic mechanisms of antinociception in inflammation: local opioid receptors and β-endorphinJ Neurosci199010129212982158530
- KhodorovaANavarroBJouavilleLSEndothelin B receptor triggers an endogenous analgesic cascade at sites of peripheral injuryNat Med200391055106112847519
- IbrahimMMPorrecaFLaiJCB2 cannabinoid receptor activation produces antinociception by stimulating peripheral release of endogenous opioidsProc Natl Acad Sci U S A20051023093309815705714
- TobinVALudwigMThe role of the actin cytoskeleton in oxytocin and vasopressin release from rat supraoptic nucleus neuronsJ Physiol20075821337134817478532
- LuJKatanoTUtaDFurueHItoSRapid S-nitrosylation of actin by NO-generating donors and in inflammatory pain model miceMol Pain2011710122192148
- HuangXChengZSuQNeuroprotection by nicotine against colchicine-induced apoptosis is mediated by PI3-kinase-Akt pathwaysInt J Neurosci201212232433222248034
- ZhengYSarrMGTranslocation of transfected GLUT2 to the apical membrane in rat intestinal IEC-6 cellsDig Dis Sci2012571203121222116644