Abstract
Celecoxib, diclofenac, ibuprofen, and nimesulide are nonsteroidal anti-inflammatory drugs (NSAIDs) very commonly used for the treatment of moderate to mild pain, together with paracetamol (acetaminophen), a very widely used analgesic with a lesser anti-inflammatory effect. In the study reported here, we tested the efficacy of celecoxib, diclofenac, and ibuprofen on preprotachykinin mRNA synthesis, substance P (SP) release, prostaglandin E2 (PGE2) release, and protein kinase C epsilon (PKCɛ) translocation in rat cultured sensory neurons from dorsal root ganglia (DRGs). The efficacy of these NSAIDs was compared with the efficacy of paracetamol and nimesulide in in vitro models of hyperalgesia (investigated previously). While nimesulide and paracetamol, as in previous experiments, decreased the percentage of cultured DRG neurons showing translocation of PKCɛ caused by 100 nM thrombin or 1 μM bradykinin in a dose-dependent manner, the other NSAIDs tested did not have a significant effect. The amount of SP released by peptidergic neurons and the expression level of preprotachykinin mRNA were assessed in basal conditions and after 70 minutes or 36 hours of stimulation with an inflammatory soup (IS) containing potassium chloride, thrombin, bradykinin, and endothelin-1. The release of SP at 70 minutes was inhibited only by nimesulide, while celecoxib and diclofenac were effective at 36 hours. The mRNA basal level of the SP precursor preprotachykinin expressed in DRG neurons was reduced only by nimesulide, while the increased levels expressed during treatment with the IS were significantly reduced by all drugs tested, with the exception of ibuprofen. All drugs were able to decrease basal and IS-stimulated PGE2 release. Our study demonstrates novel mechanisms of action of commonly used NSAIDS.
Introduction
Nimesulide, celecoxib, diclofenac, and ibuprofen are nonsteroidal anti-inflammatory drugs (NSAIDs) in general use for the treatment of mild–moderate pain, acting mostly via cyclooxygenase (COX)-1/-2 inhibition, but also via other mechanisms, only partially identified. The anti-inflammatory and analgesic actions of these drugs go beyond COX inhibition, affecting a broad range of pain and inflammatory mediators and intracellular pathways, acting in a multifactorial fashion.Citation1–Citation4 Paracetamol is largely used as an analgesic and antipyretic, which, because of its lesser anti-inflammatory activity and poorer inhibition of COX-1 and COX-2,Citation5,Citation6 has not traditionally been considered a NSAID. This is despite recent recognition of its effects as a substrate and inhibitor of the peroxidase function of COX-1 and COX-2 (which are stronger on the latter).Citation7,Citation8
In a recent paper, we proposed that nimesulide and paracetamol may exert novel mechanisms of action, likely relevant for their analgesic action, by modulating protein kinase C epsilon (PKCɛ) and substance P (SP) in the peripheral sensory neurons.Citation9 In the present paper, we extend a similar analysis to other NSAIDs in general use.
PKCɛ has a crucial role in sensitizing peripheral neurons to painful stimuli leading to the sensitization of transient receptor potential vanilloid 1 (TRPV1) and other nociceptor-specific ion channels. Translocation can be easily visualized with immunocytochemistry.Citation9–Citation13 Inhibition of translocation by nimesulide – and by paracetamol to a lesser extent – has been recently proposed by our group as a novel analgesic mechanism activated by these drugs.Citation9
SP – a neuropeptide derived from the preprotachykinin (PPT)-A gene produced in a subset of peptidergic nociceptive neurons located in the dorsal root ganglia (DRGs) and trigeminal gangliaCitation14 – is required for experiencing moderate to intense pain.Citation15 Centrally, SP is released in the superficial laminae of the spinal dorsal horn, where it participates in the transmission of noxious stimuli.Citation16 Further, when released from peripheral nerve endings, SP is an important mediator of neurogenic inflammation, together with other peptides,Citation17–Citation19 and is involved in nociceptor sensitization, hyperalgesia, and allodynia following tissue injury.Citation19,Citation20 In this paper, we report on the modulation of SP synthesis and release by widely used analgesic and anti-inflammatory drugs. In relation to this, we have previously demonstrated that nimesulide significantly reduces the synovial fluid concentrations of SP in patients with knee osteoarthritis and, more recently, that nimesulide decreases synthesis and release of SP in cultured DRG neurons.Citation9
Materials and methods
Drugs
The following anti-inflammatory/analgesic drugs were used in this study: nimesulide (Helsinn Healthcare, Lugano, Switzerland) and ibuprofen, celecoxib, diclofenac, and paracetamol (all obtained from Sigma-Aldrich, Milan, Italy).
DRG cultures of sensory neurons
Sprague Dawley rats (aged 14–21 days old) were sacrificed following total anesthesia according to European and Italian legislation, following protocols according to the guidelines of the Committee for Research and Ethical Issues of the International Association for the Study of Pain.Citation21 Experimental procedures and research project were approved by local institutional animal care and use committee. DRGs were extracted and digested with 0.125% collagenase (Worthington Biochemical, Freehold, NJ, USA) for 60 minutes at 37°C, then dissociated with gentle trituration and allowed to attach onto glass coverslips or Petri dishes pretreated with 10 μg/mL poly-l-lysine. Glass coverslips were also treated with 20 μg/mL laminin (Sigma-Aldrich). The culture medium – Dulbecco’s modified Eagle’s medium (DMEM) – was added with 1.5 μg/mL cytosine 1-d-arabinofuranoside (Sigma-Aldrich), 10% fetal bovine serum (FBS), 1% L-glutamine, 1% penicillin/streptomycin (Invitrogen, San Diego, CA, USA) as previously described.Citation22 For the immunocytochemistry experiments, the medium also contained 100 ng/mL nerve growth factor (7s; Sigma-Aldrich) to increase bradykinin (BK)- and thrombin (THR)-receptor expression.Citation12,Citation23
Immunocytochemistry
The subcellular localization of the epsilon isoform of protein kinase C (ie, PKCɛ) was investigated as previously described.Citation9,Citation13,Citation24 In brief, rat DRG neurons cultured for 2–3 days were rapidly exposed to BK (at 1 μM concentration) or THR (at 100 nM concentration) for 30 seconds, then fixed for 10 minutes at room temperature with paraformaldehyde (4% formaldehyde and 4% sucrose, dissolved in phosphate-buffered saline [PBS]/distilled water 2:1). The concentrations of BK and THR in some experiments were reduced to 10 nM to test the effects of drugs on non-saturating concentrations of stimulants inducing translocation. Dimethyl sulfoxide was used to prepare stock solutions of NSAIDs and paracetamol. The final concentration of dimethyl sulfoxide applied to cells was always lower than 1:1000. Cells were treated with 10 μM of the tested drug for 2 hours before stimulation with THR and BK. As there are pharmacokinetic differences between paracetamol and nimesulide that lead to a higher plasma concentration of free paracetamol in clinical treatment, paracetamol was also tested at 100 μM. Fixed cells were washed three times with PBS and 0.1% fish-skin gelatin, treated with Triton™ X-100 (0.2% in PBS) for 30 minutes at room temperature, and incubated at 4°C for 8–12 hours with an affinity-purified polyclonal antibody anti-PKCɛCitation10 diluted 1:1000 in PBS-T/gelatin (PBS with 0.05% Triton X-100). Coverslips were thoroughly rinsed then stained for 1 hour at room temperature with goat anti-rabbit immunoglobulin G conjugated to Alexa Fluor® 488 (1:200; Invitrogen), washed three times in PBS/gelatin, and analyzed using a confocal microscope (Leica SP2, Leica, Switzerland). Activation of PKCɛ downstream of Gq-coupled membrane receptors leads to translocation from the cytoplasm to the neuronal cell membrane (see ). Translocation was assessed by measuring fluorescence intensity along a line across the cell, avoiding the nucleus (for details, see Cesare et alCitation10). Neurons in which peak intensity at the cell membrane was at least 2.0× greater than the mean of cytoplasm intensity were considered positive.
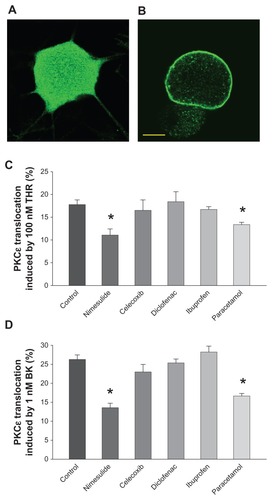
Figure 1 Protein kinase C epsilon (PKCɛ) translocation induced by bradykinin (BK) and thrombin (THR) is inhibited by nimesulide and paracetamol, but not by other nonsteroidal anti-inflammatory drugs (NSAIDs) tested. (A and B) Confocal optical sections of cultured sensory neurons treated with THR (100 nM) for 30 seconds and subsequently fixed and stained for PCKɛ with a polyclonal specific antibody: (A) typical neuron showing no response; (B) typical THR-responsive neuron with translocation of PKCɛ to the plasma membrane. Neurons treated with 1 μM BK for 30 seconds had identical appearance. Neurons not treated with BK or THR showed no sign of translocation. Scale bar 5 μm. (C) PKCɛ translocation in cultured dorsal root ganglion neurons induced by THR (100 nM for 30 seconds), quantified as the percentage of neurons showing a clear edge. Translocation induced by THR was significantly reduced in the presence of nimesulide and paracetamol, while celecoxib, diclofenac, and ibuprofen were ineffective. All drugs were applied for 2 hours at a concentration of 10 μM before treatment with THR. (D) The effects of NSAIDs and paracetamol on translocation induced by BK (1 μM for 30 seconds) were largely similar and nimesulide was significantly stronger than paracetamol (P < 0.05).
Culture stimulation and drug treatment
At Day 2/3 in vitro, cultures were stimulated using a cocktail of inflammatory/pro-algesic mediators (termed “inflammatory soup” [IS]) composed of 100 nM THR, 1 μM BK, 100 nM endothelin-1, 25 mM potassium chloride, dissolved in culture medium (DMEM + 10% FBS) as previously described.Citation9 When required, at a concentration of 10 μM, each of the drugs to be tested was pre-applied to cultures before treatment with IS for the time considered appropriate to be fully effective (see “Results”). Paracetamol was also tested, but at a concentration of 100 μM. Cells were then stimulated with IS with/without drugs for either 70 minutes or 36 hours. At the end of the incubation times, media and/or cells were separately stored and processed as described following.
Measurement of SP and prostaglandin E2 (PGE2) in culture media
SP was measured by radioimmunoassay (RIA) according to methods previously described and validated using an antibody raised in rabbit against synthetic antigen mimicking an epitope in the C terminal of SP.Citation25,Citation26 I125-SP was purchased from PerkinElmer (Monza, Italy). Culture media pH was lowered with 1 N acetic acid before the procedure. Sensitivity of the RIA was 10 pg/tube and the intra- and inter-assay variation coefficients were 8% and 11%, respectively.
Quantitative determination of PGE2 was performed by enzyme immunoassay (EIA) using a commercially available EIA kit (Cayman Chemical, Ann Arbor, MI, USA) with a sensitivity of 15 pg/mL.
RNA isolation and real-time polymerase chain reaction (PCR)
This was undertaken as outlined in Vellani et al:Citation9
Total RNA from DRG cells was purified using TRIzol reagent (Invitrogen, Life Technologies, San Giuliano Milanese, Italy). Cells were lysed directly in the culture dish, according to the manufacturer’s instructions and resuspended in 8 μL of formamide. After purification, total RNA concentrations were determined from the sample absorbance value at 260 nm. 3000 ng of total RNA were treated with DNase (DNA-free-Ambion) to avoid false-positive results due to amplification of contaminating genomic zDNA. First strand cDNA was synthesized from 1000 ng of total RNA in a final volume of 20 μL using M-MLV RT (Moloney Murine Leukemia Virus Reverse Transcriptase; Invitrogen, San Giuliano Milanese, Italy). cDNA (2 μL) was subjected to real-time quantitative PCR using ABI PRISM 7000 (Applied Biosystems, Forster City, CA). TaqMan PCR was performed in 25 μL volumes using Real Master Mix Probe ROX (Eppendorf, Hamburg, Germany). Custom probes for preprotachykinin (PPT, Genbank accession number M15191) and GAPDH [glyceraldehyde 3-phosphate dehydrogenase] (Genbank accession number AF106860) were prepared by Applied Biosystem. The probes were designed to span an intron in order to avoid potential amplification of genomic DNA in the analyzed samples.Citation9,Citation25 The probes were labeled at the 5′ end with 6-carboxy fluorescein (FAM) and at the 3′ end with 6-carboxy-tetramethyl rhodamine (TAMRA). All PCR assays were performed in duplicate. Before using the ΔΔCT method for relative quantification, we performed a validation experiment to demonstrate that the efficiencies of the two different probes (target and reference) are equal. The reaction conditions were as follows: 95°C for 2 min, followed by 40 cycles at 95°C for 15 s (denaturation) and 60°C for 1 min (annealing and elongation). As controls, we used the reaction mixture without the cDNA. Threshold cycle numbers (CT) were determined with an ABI PRISM 7000 Sequence Detection System (version 1.1 software) and transformed using the ΔCT (2−ΔΔCT) comparative method. Gene-specific expression values were normalized to expression values of GAPDH (endogenous control) within each sample. The levels of preprotachykinin were expressed relative to the calibrator value control cells. Relative quantification was performed using the comparative method. The amount of target, normalized to an endogenous reference and relative to a calibrator, is given by 2−ΔΔCT. Briefly, the ΔCT value is determined by subtracting the average GAPDH CT value from the average PPT CT in the same sample. The calculation of ΔΔCT involves subtraction of the ΔCT calibrator value.
Statistical analysis
Data were analyzed by analysis of variance followed by Bonferroni’s t-test for multiple comparisons. An effect was considered significant if the P value was <0.05.
Results
Modulation of PKCɛ translocation
In sensory neurons, the activation of PKCɛ by inflammatory mediators (or, artificially, by phorbol esters) is followed by its translocation to the plasma membrane. Translocation occurs rapidly and can be directly observed with immunocytochemistry ().Citation9,Citation12,Citation13,Citation23 In this study, PKCɛ activation was quantified as the percentage of neurons showing membrane translocation. This method allowed for the measurement of reproducible time-course and dose–response curves, as previously shown.Citation12,Citation13 Following exposure to 100 nM THR or 1 μM BK, sufficient to saturate the specific receptors, maximum translocation was always observed.Citation10,Citation11 Application times longer than 30 seconds would allow PKCɛ to become internalized into perinuclear vesicles, decreasing the number of translocation-positive neurons, as has been shown previously.Citation10,Citation11,Citation13 As maximum percentage of translocation was reliably measured at 30 seconds of exposure to the agonist concentrations mentioned, as in previous studies,Citation9 these parameters were used for most of our work, with the exception of some experiments with nimesulide and paracetamol, in which we tested a roughly half-maximal agonist concentration (10 nM applied for 30 seconds in the case of both stimulants).Citation10,Citation11,Citation13 THR applied on cultures for 30 seconds at 100 nM and BK applied on cultures for 30 seconds at 1 μM, triggered translocation in 17.8% ± 1.0% and 26.3% ± 1.2% of DRG neurons, respectively, in this set of experiments, a percentage similar to that previously obtained;Citation9 THR and BK applied for 30 seconds at 10 nM triggered translocation in 8.8% ± 0.9% and in 14.4% ± 0.7% of DRG neurons, respectively.
As shown in , when compared with neurons pretreated with vehicle solution, the percentage of neurons in which THR (A) and BK (B) induced translocation was significantly decreased to 11.1% ± 1.4% and 13.6% ± 1.2%, respectively, by a 2-hour pre-application of 10 μM nimesulide (P < 0.001). This was similar to the decrease obtained in previous work,Citation9 in which a different set of experiments was used. Therefore, nimesulide reduced the translocation induced by saturated concentrations of THR and BK by ~38% and ~48%, respectively. This percentage was largely similar to that achieved in experiments in which 10 μM nimesulide was pre-applied onto neurons stimulated with 10 nM THR and BK, which produced translocation in 5.3% ± 0.8% and 8.2% ± 0.5% of neurons, respectively (about ~40% and 43% reduction, respectively). In contrast, the application of the NSAIDs celecoxib, diclofenac, and ibuprofen (all at 10 μM concentration, also pre-applied for 2 hours) on THR- and BK-induced translocation did not cause a significant variation when compared with control experiments. Paracetamol (10 μM), also as previously observed,Citation9 inhibited translocation by 100 nM THR and 1 μM BK to 13.4% ± 0.5% and 16.6% ± 0.7%, respectively, with a reduction of ~25% and ~37%, respectively. A larger concentration of paracetamol (100 μM) was tested on translocation by 100 nM THR and 1 μM BK causing a reduction of ~27% and ~34%, respectively, versus the respective controls. We concluded that the maximal paracetamol effect on translocation was achieved at a concentration of 10 μM.
The reduction in translocation-positive neurons caused by nimesulide and paracetamol was not accompanied by an increase in neurons showing PKCɛ internalization after translocation, as such neurons comprised less than 1% of the total number of translocated neurons, both in the control and drug-treated neurons. All experiments were repeated between six and twelve times.
Longer applications of NSAIDs or paracetamol at 10 μM (increased from 2 hours to overnight) before stimulation with THR or BK had no significant effect (results not shown).
Effects of drugs on PPT mRNA synthesis
SP is derived from its precursor, PPT. PPT mRNA levels were assessed in DRG cultures with real-time PCR, as shown in , in which measurements are expressed as relative mRNA levels of PPT normalized to GAPDH. Gene specific expression values are normalized to GAPDH (endogenous control) within each sample (ΔCT). Each ΔCT is transformed using the ΔCT (2−ΔΔCT) comparative method. In this way expression values of control cultures are arbitrary considered as 1, and treatment values are expressed as fold increase in relation to control. In basal conditions approximately 60 hours after plating, measurable PPT mRNA levels could be detected. Basal amounts of PPT mRNA were significantly decreased by 36-hours’ exposure to nimesulide, while they remained unchanged after 36-hours of treatment with other tested NSAIDs and paracetamol (all at 10 μM).
Figure 2 Modulation of basal and induced preprotachykinin (PPT) mRNA expression in cultured rat dorsal root ganglion cells by nonsteroidal anti-inflammatory drugs (NSAIDs) and paracetamol. (A) Basal expression of PPT mRNA was significantly reduced by nimesulide but was unchanged by treatment with other NSAIDs or paracetamol (all at 10 μM). (B) Nimesulide, paracetamol, celecoxib, and diclofenac significantly decreased upregulation of PPT mRNA expression induced by the inflammatory soup, while ibuprofen was ineffective.
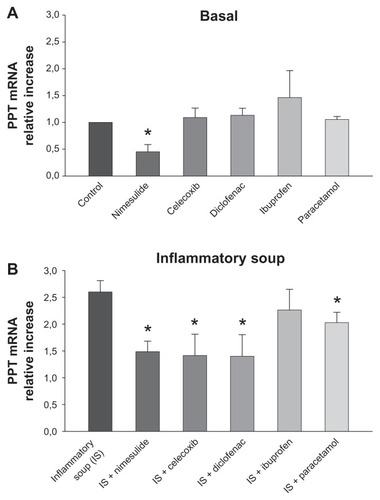
Stimulation with IS raised mRNA amounts by approximately 2.6-fold when compared with the control (), consistent with previous similar experiments.Citation9 Drug treatments (all at 10 μM) were added on cells 10 minutes before stimulation with IS and maintained in culture for 36 hours. Nimesulide, celecoxib, and diclofenac significantly decreased the increase of PPT mRNA expression induced by the IS with similar levels of efficacy, and paracetamol was somewhat less, although still significantly, effective. In contrast, ibuprofen had no effect.
Effects on basal and stimulated SP release
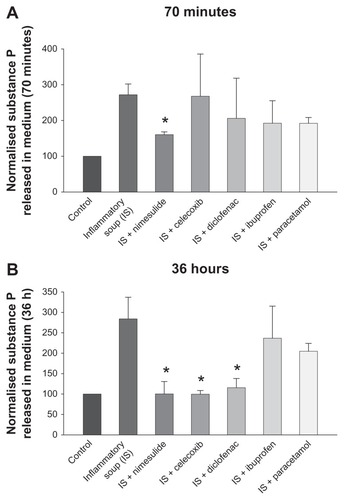
Concentrations of SP released by cultured sensory neurons in culture medium were measured by RIA (see “Materials and methods”). After 24 hours in culture, individual coverslips from the same cultures were treated either with vehicle or IS. Separate IS-treated coverslips contained either one of the NSAIDs investigated, paracetamol (all at 10 μM) or vehicle solution, giving a total of seven conditions. Coverslips were pretreated with the same concentration of the drugs investigated for 10 minutes before addition of IS, and coverslips containing only IS were pretreated for the same time with vehicle medium. Thus, in these experiments, the effect of the drugs was tested on the IS-stimulated release of SP and not on the basal release which was not modified by nimesulide or paracetamol (see Vellani et alCitation9) or any of the drugs tested in this paper (data not shown). In , all results shown are normalized to basal (vehicle-only) levels. The treatments lasted for either 70 minutes or 36 hours (, respectively), then the medium was collected and stored at −80°C before SP quantitation. At 70 minutes after exposure to the IS, nimesulide was the only compound found to be capable of significantly reducing SP release. At 36 hours following exposure to the IS, nimesulide, celecoxib, and diclofenac significantly reduced the IS-induced release of SP, while reductions caused by paracetamol and ibuprofen were not statistically significant. Paracetamol was also tested at a concentration of 100 μM; at this concentration, paracetamol did not significantly reduce stimulated SP release in medium at 70 minutes or 36 hours (n = 4, data not shown).
Figure 3 Modulation by nonsteroidal anti-inflammatory drugs and paracetamol of substance P (SP) release from dorsal root ganglion neurons after 70 minutes (A) and 36 hours (B) of treatment with inflammatory soup.
Effects on PGE2 release in medium
PGE2 was measured in cultures of sensory neurons by EIA.Citation25,Citation27 Little, if any, PGE2 was measured in the medium, the only traces being present in the FBS added to the DMEM (see “Materials and methods”). After 24 hours in culture, the medium was removed and the cells were treated either with vehicle or IS, with or without NSAIDs or paracetamol (all at 10 μM). At 70 minutes or 36 hours of treatment, the medium was removed from the cultures and the PGE2 released was measured. The PGE2 released by unstimulated DRG cultures in this set of experiments was 143 ± 22.8 pg/mL after 70 minutes and 492 ± 18.6 pg/mL after 36 hours. PGE2 amounts significantly increased in comparison with the control by 70 minutes’ or 36 hours’ treatment with the IS: compared with the control, 70 minutes’ stimulation augmented PGE2 concentrations by about 2.3-fold, while the increase was about 10.5-fold after 36 hours (see ).
Figure 4 Effect of nonsteroidal anti-inflammatory drugs and paracetamol on release of prostaglandin E2 (PGE2) from dorsal root ganglion neurons. (A and C) Effects of drugs on basal PGE2 levels in cultures incubated for 70 minutes or 36 hours. (B and D) Effects of drugs on PGE2 levels following 70 minutes (B) and 36 hours (D) of treatment with inflammatory soup. Note that the scales in panels (C and D) are identical in the range 0–500 pg/mL, allowing direct comparison of data.
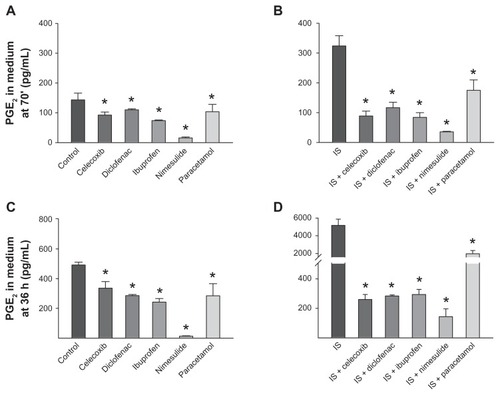
Treatment with NSAIDs (10 μM; applied for 70 minutes) strongly inhibited both basal () and IS-stimulated PGE2 levels () released in the medium compared with untreated cells, and not only inhibited the IS effect, but also reduced the amount of PGE2 released to concentrations even lower than the basal level (). Similarly, 36 hours’ treatment with all NSAIDs reduced basal and stimulated PGE2 down to levels lower than those present in the medium in unstimulated cultures ().
Paracetamol at 10 μM had significant effects on basal and IS-induced PGE2 release at both 70 minutes and 36 hours. However, the inhibition of IS-induced PGE2 release, in particular at 36 hours, was only partial, as PGE2 was higher than in cells that had not received IS treatment. Paracetamol at 100 μM also exerted only partial inhibition on basal and IS-induced PGE2 release, both at 70 minutes and 36 hours. Basal PGE2 release was reduced by ~28% and ~42% at 70 minutes and 36 hours, respectively, by 10 μM paracetamol (see ), and by ~54% and ~78%, respectively, by 100 μM paracetamol. IS-stimulated PGE2 release was inhibited by ~46% and ~62% by 10 μM paracetamol at 70 minutes and 36 hours, respectively, and by ~35% and ~62% by 100 μM paracetamol at 70 minutes and 36 hours, respectively.
Discussion
The importance of PKCɛ activation in sensory neurons in peripheral mechanisms of hyperalgesia and allodynia has been studied in many papers and largely agreed upon. Cesare et alCitation10 first showed that the increase (sensitization) of the heat-gated current induced by BK in cultured nociceptors is due to PKCɛ and, in the following years, a large number of studies validated the important role of this isoform in inflammatory hyperalgesia and allodynia in cellular and behavioral models.Citation22,Citation28–Citation30 PKCɛ is highly expressed in small-diameter sensory neurons, in which it is essential for chronic hyperexcitability; nevertheless, when blocked, it has only negligible effects on other sensory or non-sensory functions.Citation30 Therefore, PKCɛ is currently considered a well-validated mediator of chronic pain, both in inflammatory and neuropathic models,Citation31,Citation32 and as a new and potentially important target for therapeutic treatment.
In this context, the reported inhibition of PKCɛ translocation by nimesulide and paracetamol (previously reported for both drugs at 10 μMCitation9 and in the present paper by paracetamol at 100 μM) appears to be highly relevant to properly understand the pharmacological actions of these drugs. However, it is interesting to notice that, as shown in , not all NSAIDs inhibit PKCɛ: celecoxib, diclofenac, and ibuprofen did not interfere with translocation by BK or THR, therefore their analgesic actions cannot be due to any effect on translocation.
Prostaglandins (PGs) are normally produced in large quantities after the exposure of cultured sensory neurons to BK, THR, and IS. They can be produced not only by nociceptive neurons themselves, which express COX-1,Citation12,Citation33 but also by satellite cells and Schwann cells, which are present in large numbers together with neurons in DRG cultures. These cells express most receptors for the same inflammatory mediators expressed by neurons, and release PGs and many other mediators.Citation34,Citation35 In principle, inhibition of PG synthesis, in particular of PGE2, which can activate PKCɛ,Citation24 might be responsible for the effect of NSAIDs, as previously hypothesized.Citation9 However, in this study, we demonstrated that the other NSAIDs at the concentrations tested – which, like nimesulide, would strongly inhibit COX () – did not inhibit translocation. Therefore, an indirect effect on translocation via COX inhibition is unlikely, and some more specific inhibitory mechanisms on translocation by nimesulide and paracetamol, in comparison to other NSAIDs, must be responsible for the effects observed.
Nimesulide’s and paracetamol’s inhibitory effects are basically identical both on BK- and THR-induced translocation, suggesting that these drugs affect other mechanisms of the intracellular pathway leading to translocation rather than specifically interacting with BK and THR membrane receptors – possibly directly PKCɛ itself or the interaction of PKCɛ with its specific receptors for activated PKC.Citation36
SP is one of the mediators of inflammatory pain due to a complex pattern of local changes, including the synthesis and release of many pro-nociceptive and pro-inflammatory mediators. These mediators lower nociceptive thresholds and increase neuronal membrane excitability, leading to hyperalgesia and allodynia.Citation37,Citation38 Following noxious stimulation, SP is exocitosed through a very complex mechanism involving several important events including calcium inflow, 1-4-5 inositol triphosphate-mediated intracellular calcium release, extracellular signal-regulated kinases, protein kinase A, and COX activation and release of PGs. In the central nervous system, SP has an important function in the sensitization of spinal neurons, while, in the peripheral nervous system, it produces vasodilatation, decreases nociceptive threshold, and participates in neurogenic inflammation.Citation17 In the immune system, SP can directly induce the chemotaxis of macrophages and monocytes and the synthesis of several pro-inflammatory cytokines.Citation26,Citation39,Citation40 These mechanisms play a role in the diffusion of sensitization leading to secondary hyperalgesia. Therefore, modulation of spinal and peripheral SP extracellular levels appears to be a crucial control factor for the pain threshold in inflammatory hyperalgesia.
In our experiments, the increased PPT synthesis activated by IS was significantly reduced by the NSAIDs nimesulide, diclofenac, and celecoxib, but not by ibuprofen or paracetamol, while unstimulated level of synthesis was significantly affected only by nimesulide ().
Moreover, modulation of SP release is not part of the effects of paracetamol and ibuprofen. In the case of the latter, this suggests that if indirect mechanisms occur, they may not involve COX inhibition, as ibuprofen is a potent general COX inhibitor, as shown in .
Considering the models used in our experiment and the rapidity of the effect, the inhibition of SP release by nimesulide is probably caused by a direct effect on nociceptive neurons. In contrast, the inhibition of SP release by celecoxib and diclofenac took longer to occur, thus may involve indirect mechanisms, one of which may be their well-known COX inhibition, which leads to lower PG production. However, the observation that ibuprofen is unable to affect SP seems to rule out the possibility that PGs could be the main mediators involved in the effects. In addition, we previously demonstrated that paracetamol is also effective in reducing PG synthesis in DRG cultures, while being only partially able to modulate SP.Citation9 However, a main pharmacological difference between ibuprofen and the other NSAIDs studied is that ibuprofen has a higher specificity for COX-1. Since both COX-1 and COX-2 are active in DRG sensory neurons as well as in satellite cells and other cells of inflamed tissues, further studies are needed to understand the role of the two enzyme isoforms. Nevertheless, other indirect mechanisms activated by other intermediate mediators cannot be ruled out.
It has been recently reported that TRPV1 channel activity (and subsequent calcium inflow) is a potent trigger of SP production and release.Citation41 As TRPV1 sensitization is caused by PKCɛ-dependent phosphorylation, the likelihood that NSAIDs may decrease SP via PKCɛ translocation inhibition can be conceived. Conversely, the binding of SP to neurokinin 1 receptors in nociceptors sensitizes TRPV1 via PKCɛ activation.Citation42 Therefore, a positive feedback mechanism can be hypothesized, in which SP, PGs, TRPV1, and PKCɛ act together to produce hyperalgesia during inflammation.
However, one cannot exclude the possibility that the inhibition of translocation and the reduction in SP release are independent events controlled by unrelated mechanisms. In any case, the effects of celecoxib, diclofenac, and nimesulide on numerous important related mediators can be useful not only to control acute hyperalgesia but also to avoid its progression to chronic pain.
Conclusion
Our data help to clarify the range of pharmacological effects of widely used NSAIDs and of paracetamol in their use as analgesics. We are aware that it is not possible to claim that the effects described would occur to a quantitatively similar extent in human patients. In fact, considering the pharmacokinetics, in clinical treatment, the plasma concentrations of free drug would possibly be lower than the ones used in this study (as well as in other studies) in vitro, as the aim was to show the maximum effect obtainable in our in vitro models. Nevertheless, our results suggest that NSAIDs generally and nimesulide in particular are drugs with multifactorial modes of action with an increasing number of novel and interesting targets.
Acknowledgments
Work was funded by the Fondazione Cassa di Risparmio di Modena and Fondazione Cassa di Risparmio di Carpi, and by an unrestricted research grant by Helsinn Healthcare SA (Lugano, Switzerland). The equipment used for automated immunocytochemistry protocols was kindly gifted by CV Scientific (Modena, Italy).
Disclosure
The authors declare no conflicts of interest in this work.
References
- RainsfordKDNimesulide – a multifactorial approach to inflammation and pain: scientific and clinical consensusCurr Med Res Opin20062261161117016846549
- BennettAVillaGNimesulide: an NSAID that preferentially inhibits COX-2, and has various unique pharmacological activitiesExpert Opin Pharmacother20001227728611249549
- WarnerTDGiulianoFVojnovicIBukasaAMitchellJAVaneJRNonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: a full in vitro analysisProc Natl Acad Sci U S A199996137563756810377455
- SuleymanHCadirciEAlbayrakAHaliciZNimesulide is a selective COX-2 inhibitory, atypical non-steroidal anti-inflammatory drugCurr Med Chem200815327828318333314
- HarvisonPJEganRWGalePHNelsonSDAcetaminophen as a cosubstrate and inhibitor of prostaglandin H synthaseAdv Exp Med Biol19861977397473094341
- OhkiSOginoNYamamotoSHayaishiOProstaglandin hydroperoxidase, an integral part of prostaglandin endoperoxide synthetase from bovine vesicular gland microsomesJ Biol Chem19792543829836104998
- BoutaudOAronoffDMRichardsonJHMarnettLJOatesJADeterminants of the cellular specificity of acetaminophen as an inhibitor of prostaglandin H(2) synthasesProc Natl Acad Sci U S A200299107130713512011469
- GrahamGGScottKFMechanism of action of paracetamolAm J Ther2005121465515662292
- VellaniVFranchiSPrandiniMNimesulide inhibits protein kinase C epsilon and substance P in sensory neurons – comparison with paracetamolJ Pain Res2011417718721811393
- CesarePDekkerLVSardiniAParkerPJMcNaughtonPASpecific involvement of PKC-epsilon in sensitization of the neuronal response to painful heatNeuron199923361762410433272
- VellaniVKinseyAMPrandiniMProtease activated receptors 1 and 4 sensitize TRPV1 in nociceptive neuronesMol Pain201066120875131
- VellaniVPrandiniMGiacomoniCPavesiGRavegnaniLMagheriniPCFunctional endothelin receptors are selectively expressed in isolectin B4-negative sensory neurons and are upregulated in isolectin B4-positive neurons by neurturin and glia-derived neurotropic factorBrain Res20111381313721241671
- VellaniVColucciMLattanziRSensitization of transient receptor potential vanilloid 1 by the prokineticin receptor agonist Bv8J Neurosci2006265109511616687502
- SacerdotePLevriniLPeripheral mechanisms of dental pain: the role of substance PMediators Inflamm2012201295192022474402
- WoolfCJMannionRJNeumannSNull mutations lacking substance: elucidating pain mechanisms by genetic pharmacologyNeuron1998206106310669655494
- CaoYQMantyhPWCarlsonEJGillespieAMEpsteinCJBasbaumAIPrimary afferent tachykinins are required to experience moderate to intense painNature199839266743903949537322
- MaggiCATachykinins and calcitonin gene-related peptide (CGRP) as co-transmitters released from peripheral endings of sensory nervesProg Neurobiol19954511987716258
- WhiteDMRelease of substance P from peripheral sensory nerve terminalsJ Peripher Nerv Syst19972319120110975725
- TangHBLiYSArihiroKNakataYActivation of the neurokinin-1 receptor by substance P triggers the release of substance P from cultured adult rat dorsal root ganglion neuronsMol Pain200734218157919
- SuzukiRMorcuendeSWebberMHuntSPDickensonAHSuperficial NK1-expressing neurons control spinal excitability through activation of descending pathwaysNat Neurosci20025121319132612402039
- ZimmermanMEthical guidelines for investigations of experimental pain in conscious animals1983162109110
- VellaniVMapplebeckSMoriondoADavisJBMcNaughtonPAProtein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamideJ Physiol2001534Pt 381382511483711
- LeeYJZachrissonOTongeDAMcNaughtonPAUpregulation of bradykinin B2 receptor expression by neurotrophic factors and nerve injury in mouse sensory neuronsMol Cell Neurosci200219218620011860272
- VellaniVZachrissonOMcNaughtonPAFunctional bradykinin B1 receptors are expressed in nociceptive neurones and are upregulated by the neurotrophin GDNFJ Physiol2004560Pt 239140115319421
- BianchiMFranchiSFerrarioPEffects of the bisphosphonate ibandronate on hyperalgesia, substance P, and cytokine levels in a rat model of persistent inflammatory painEur J Pain200812328429217664076
- BianchiMMartucciCBiellaGFerrarioPSacerdotePIncreased substance P and tumor necrosis factor-alpha level in the paws following formalin injection in rat tailBrain Res200410191–225525815306260
- BianchiMMartucciCFerrarioPFranchiSSacerdotePIncreased tumor necrosis factor-alpha and prostaglandin E2 concentrations in the cerebrospinal fluid of rats with inflammatory hyperalgesia: the effects of analgesic drugsAnesth Analg2007104494995417377112
- HuangJJZhangXMMcNaughtonPAModulation of temperature- sensitive TRP channelsSemin Cell Dev Biol200617663864517185012
- AleyKOMessingROMochly-RosenDLevineJDChronic hypersensitivity for inflammatory nociceptor sensitization mediated by the epsilon isozyme of protein kinase CJ Neurosci200020124680468510844037
- ReichlingDBLevineJDCritical role of nociceptor plasticity in chronic painTrends Neurosci2009321261161819781793
- SouroujonMCMochly-RosenDPeptide modulators of protein-protein interactions in intracellular signalingNat Biotechnol199816109199249788346
- BrandmanRDisatnikMHChurchillEMochly-RosenDPeptides derived from the C2 domain of protein kinase C epsilon (epsilon PKC) modulate epsilon PKC activity and identify potential protein-protein interaction surfacesJ Biol Chem200728264113412317142835
- ChopraBGiblettSLittleJGCyclooxygenase-1 is a marker for a subpopulation of putative nociceptive neurons in rat dorsal root gangliaEur J Neurosci200012391192010762321
- VellaniVPetrosinoSDe PetrocellisLFunctional lipidomics. Calcium-independent activation of endocannabinoid/endovanilloid lipid signalling in sensory neurons by protein kinases C and A and thrombinNeuropharmacology20085581274127918329052
- CampanaWMSchwann cells: activated peripheral glia and their role in neuropathic painBrain Behav Immun200721552252717321718
- Mochly-RosenDLocalization of protein kinases by anchoring proteins: a theme in signal transductionScience199526852082472517716516
- WoolfCJAllchorneASafieh-GarabedianBPooleSCytokines, nerve growth factor and inflammatory hyperalgesia: the contribution of tumour necrosis factor alphaBr J Pharmacol199712134174249179382
- CunhaTMVerriWAJrSilvaJSPooleSCunhaFQFerreiraSHA cascade of cytokines mediates mechanical inflammatory hypernociception in miceProc Natl Acad Sci U S A200510251755176015665080
- BianchiMBrogginiMBalzariniPEffects of tramadol on synovial fluid concentrations of substance P and interleukin-6 in patients with knee osteoarthritis: comparison with paracetamolInt Immunopharmacol2003313–141901190814636839
- DelgadoAVMcManusATChambersJPProduction of tumor necrosis factor-alpha, interleukin 1-beta, interleukin 2, and interleukin 6 by rat leukocyte subpopulations after exposure to substance PNeuropeptides200337635536114698678
- TangHBNakataYThe activation of transient receptor potential vanilloid receptor subtype 1 by capsaicin without extracellular Ca2+ is involved in the mechanism of distinct substance P release in cultured rat dorsal root ganglion neuronsNaunyn Schmiedebergs Arch Pharmacol20083774–632533218034335
- ZhangHCangCLKawasakiYNeurokinin-1 receptor enhances TRPV1 activity in primary sensory neurons via PKCepsilon: a novel pathway for heat hyperalgesiaJ Neurosci20072744120671207717978048