
Abstract
Purpose
To assess the safety and efficacy of the serotonin–norepinephrine reuptake inhibitor desvenlafaxine in adults with painful diabetic peripheral neuropathy (DPN).
ClinicalTrials.gov identifiers
NCT00283842, NCT01050218.
Patients and methods
This was a 13-week, randomized, double-blind, placebo-controlled, fixed-dose study of desvenlafaxine in adults with painful DPN. The primary efficacy endpoint was change from baseline in numeric rating scale (NRS) score. Patients who completed the 13-week trial could continue in a 9-month open-label, flexible-dose extension study.
Results
A total of 412 patients were randomized to treatment with placebo or desvenlafaxine 50, 100, 200, or 400 mg/day. Of those, 240 patients continued in the extension study. After a planned interim analysis, conducted when the first 225 patients had completed 6 weeks of treatment in the short-term study, randomization to the 50 mg or 400 mg doses was stopped. At week 13, the mean change from baseline in NRS score was significantly greater compared with placebo in the desvenlafaxine 200 mg (difference [95% confidence interval {CI}]: 1.10 [0.50 to 1.70]; P<0.001) and 400 mg groups (0.91 [95% CI: 0.23 to 1.59]; P=0.027); differences from placebo were not statistically significant for the 50 mg (0.58 [95% CI: −0.08 to 1.25]) and 100 mg (0.59 [95% CI: –0.03 to 1.21]) groups. Nausea and dizziness were the most common treatment-emergent adverse events reported in the short-term study, and the most common adverse events leading to discontinuation in the short-term study and the extension. Adverse events rates were dose-dependent in the short-term studies.
Conclusion
Desvenlafaxine was effective in relieving pain associated with DPN at doses of 200 and 400 mg/day, and improved activity impairment at all doses assessed. Desvenlafaxine was generally well-tolerated in the short-term and long-term studies.
Introduction
Painful manifestations of diabetic peripheral neuropathy (DPN) affect between 10% and 26% of patients with diabetes mellitus and are associated with substantial morbidity and higher rates of premature mortality.Citation1,Citation2 Tricyclic antidepressants (TCAs) and opioids are effective treatments for the painful symptoms of DPN,Citation3 but side effects may limit their use for long-term pain management.Citation4–Citation6 Currently in the US, only two agents, the alpha-2-delta calcium channel agonist pregabalinCitation7 and the serotonin–norepinephrine reuptake inhibitor (SNRI) duloxetineCitation8 are indicated for the treatment of pain associated with DPN. The two agents have demonstrated efficacyCitation9 with improved tolerability compared to TCAs and opioids.Citation10–Citation12
Although the pathophysiology underlying chronic neuropathic pain is not fully understood, both peripheral and central mechanisms are thought to be involved.Citation13 Serotonergic and noradrenergic systems are known to play a role in descending pain inhibitory pathways,Citation2 and the analgesic effects of serotonergic and noradrenergic agents, including TCAs and SNRIs, are thought to be attributable to the modulation of those norepinephrine and serotonin inputs.Citation14
Desvenlafaxine (administered as desvenlafaxine succinate) is a potent and selective SNRICitation15 that has established efficacy in the treatment of major depressive disorder (MDD), with 50 mg/day as the recommended therapeutic dose.Citation16–Citation18 Desvenlafaxine is the major active metabolite of venlafaxine, and is approved in the US and other countries for treatment of MDD in adults.Citation19 The parent compound, venlafaxine, was found to be a safe and effective analgesic in a 6-week, double-blind, randomized placebo-controlled trial in metabolically stable patients with painful diabetic nephropathy.Citation20 In that study, patients who received 6-week treatment with venlafaxine 150–225 mg/day achieved a significantly greater reduction from baseline in mean Visual Analog Pain Intensity (VAS-PI) score compared with placebo-treated patients (50% versus 27% reduction, respectively, at week 6; P<0.001), and the percentage of patients with at least a 50% reduction from baseline on the VAS-PI was significantly greater for venlafaxine (56%) compared to placebo (34%; P<0.01).
Based on the efficacy of other SNRIs for the treatment of DPN, two studies were conducted to assess the safety and efficacy of desvenlafaxine in patients with DPN as part of a development program for treatment of pain. The two studies were designed to test the hypothesis that desvenlafaxine would reduce neuropathic pain associated with DPN, as measured by the numeric rating scaleCitation21 (NRS) score. The primary objective of the first, a 13-week, randomized, double-blind, placebo-controlled study, was to assess the safety and efficacy of four fixed oral doses (50, 100, 200, and 400 mg) of desvenlafaxine in adult outpatients with neuropathic pain associated with DPN. The objective of the second trial, a 9-month open-label extension study, was to evaluate the long-term safety of desvenlafaxine in patients who had completed the double-blind, placebo-controlled study. The aim of the current report is to summarize the findings of these two desvenlafaxine studies in DPN.
Methods
Study design
A Phase III, multicenter, randomized, double-blind, placebo-controlled, 13-week, adaptive-design, parallel-group study was conducted from March 2006 through June 2008 at 51 centers in the US. Following a screening period of 7–28 days, eligible patients were randomly assigned to one of five treatment groups (four doses of desvenlafaxine and placebo). The double-blind phase comprised a titration period (up to 1 week) and a maintenance period (12 weeks). Doses were tapered over a 2-week period at study completion or at patient withdrawal. An interim analysis was planned to assess the efficacy and tolerability of the desvenlafaxine doses early in the study so that randomization to doses that were not effective or tolerable would be stopped. The interim analysis was conducted after approximately half of the randomized patients had completed 6 weeks of treatment.
Eligible patients who completed the short-term study were offered the opportunity to continue in a second trial that was a 9-month open-label, flexible-dose extension study conducted from July 2006 through January 2009.
The study protocol and subsequent amendments for both trials received institutional review board or independent ethics committee approval, and the studies were conducted in accordance with the International Conference on Harmonization Guideline for Good Clinical Practice and the ethical principles that have their origin in the Declaration of Helsinki. Written informed consent was obtained from all participants before their enrollment.
Patients
Male and nonpregnant or nonlactating female outpatients aged 18 years or older were eligible for inclusion in the short-term study if they had a diagnosis of diabetes mellitus (type I or II), had written documentation of stable and optimized glycemic control for at least 3 months before randomization, and were considered unlikely to require a change in management of diabetes mellitus during study. Patients were required to have clinically and/or neurophysiologically diagnosed painful diabetic distal symmetric sensorimotor polyneuropathy affecting primarily the lower extremities. Patients were also required to have had symptoms that included chronic paresthesias, dysesthesias, hyperesthesia, hyperalgesia, or allodynia or some combination of these symptoms in the lower extremities for more than 6 months, and a score of 3 or greater on the physical examination portion of the Michigan Neuropathy Screening InstrumentCitation22 at the screening and baseline evaluations. Eligible participants had an average pain score of at least 4 (where 0= no pain and 10= worst possible pain) on the NRS for symmetrical neuropathic pain in the feet and legs, based on the last seven daily scores recorded before randomization.
Patients were excluded from study participation if they had received any previous treatment with desvenlafaxine or previous treatment with venlafaxine that could not be tolerated, had a history of drug allergies that the investigator believed would put the patient at risk, had significant asymmetrical neuropathic signs and symptoms or a neuropathy that was not due to diabetes, had other pain or any condition that may have confounded interpretation of symptoms in the lower leg and/or feet, or had suffered foot ulcers or amputation affecting all or part of a foot or toes. Patients with any of the following were ineligible: peripheral vascular disease manifested by ischemic claudication; MDD; evidence of significant risk of suicide or self-harm; uncontrolled hypertension; symptoms of orthostatic hypotension; raised intraocular pressure; elevated total cholesterol or triglycerides; unstable renal disease (creatinine clearance <50 mL/minute); gastrointestinal disease or surgery known to interfere with the absorption or excretion of drugs; current major illness or clinically important medical disease that might put the patient at risk during the study; history of any of the following: seizure disorder; neoplastic disorder within 5 years; myocardial infarction within 6 months; stroke or transient ischemic attack within 3 years; narrow angle glaucoma; or clinically important abnormalities on screening physical examination, electrocardiogram, laboratory evaluation, or urine drug screen. Any drugs used to treat the symptoms of DPN were prohibited, including (but not limited to) anticonvulsants, antidepressants, opioids, tramadol, amantadine, mexiletine, ketamine, products containing dextromethorphan, chronic daily use of analgesics, investigational drugs or procedures, and capsaicin.
Patients were eligible to enter the open-label, long-term study if they had participated in the short-term trial and continued to meet the eligibility requirements. Patients were excluded if they had any new and/or clinically important medical condition that might compromise patient safety (including, but not limited to, significant changes in glycemic control).
Treatments
Blinding of all patients and site personnel to treatment allocation was ensured by using a computerized randomization/enrollment system to assign subject numbers and study drug package numbers. Patients were randomly assigned to receive placebo or one of four fixed doses of desvenlafaxine: 50, 100, 200, and 400 mg. Patients assigned to the desvenlafaxine 400 mg group received desvenlafaxine100 mg/day for 3 days, followed by 200 mg/day for 4 days prior to receiving the maintenance dose of 400 mg/day. Patients assigned to the desvenlafaxine 200 mg group received desvenlafaxine 100 mg/day for 3 days prior to the maintenance dose of 200 mg/day. Patients assigned to the desvenlafaxine 50 mg and 100 mg groups received the assigned dose from study day 1.
At study end or at the time of withdrawal, the desvenlafaxine 400 mg dose was tapered first to 200 mg/day for 1 week, followed by up to 1 week at 100 mg/day. Patients assigned to the desvenlafaxine 200 mg group received 100 mg/day for 1 week, followed by placebo for up to 1 week. Patients assigned to the desvenlafaxine 50 mg and 100 mg groups continued to receive the assigned dose for 1 week followed by placebo for 1 week. Patients who continued into the extension study received 1 week at the double-blind taper dose prior to enrollment in the extension study.
All patients who continued in the extension study received open-label desvenlafaxine 100 mg/day for the first 2 weeks of the study. Starting on day 15 of the extension study, the dose could be increased to 200 mg/day or 400 mg/day to improve efficacy if clinically indicated. Subsequent to the interim analysis of the short-term study, titration to the 400 mg dose was discontinued. However, patients whose doses were increased to 400 mg/day prior to the interim analysis were permitted to continue receiving that dose if there were no tolerability concerns. Dose was readjusted downward to 100 mg/day for patients who were unable to tolerate the 200 mg/day dose. Patients who were unable to tolerate the 100 mg/day dose were withdrawn. Upon study completion or early withdrawal from the extension study, treatment was tapered according to a regimen identical to that used in the short-term study.
Efficacy assessments
Short-term, double-blind study
The primary efficacy endpoint in the short-term study was change from baseline in the mean NRS pain score at week 13. Patients reported “overall pain over the last 24 hours” daily using the NRS scale (0 = no pain to 10 = worst possible pain). Patients were instructed to record entries in an electronic diary at approximately the same time every day from the screening visit through study end. For the NRS pain score, the on-therapy period was divided into 1-week intervals. The weekly mean NRS pain score was calculated as the mean of daily pain scores recorded in that interval. The mean NRS score was derived at baseline, weekly, and at study end (week 13).
Secondary efficacy endpoints included Patient Global Impression of Change (PGI-C; 1= very much improved to 7= very much worse) and Clinical Global Impression of Change (CGI-C; 1= very much improved to 7= very much worse) at week 13 and change from baseline to week 13 on the sleep interference scale (SIS; 0= pain doesn’t interfere with sleep to 10= pain completely interferes with sleep), patient global symptom rating (PGSR; 0= none to 4= severe), and physician global rating (PGR; 0= none to 4= severe). Data for each of these measures were collected at baseline (except PGI-C and CGI-C) and at weeks 6 and 13. The proportion of patients with a treatment response, defined as 50% or greater decrease from baseline, was determined based on weekly NRS scores.
Health outcomes assessments were performed by evaluating the change from baseline at week 13 in several self-rated scales, including the Short Form-36 (SF-36) Health Index,Citation23 Profile of Mood States (POMS),Citation24 EuroQol Utility Assessment (EQ-5D),Citation25 Work Productivity and Activity Impairment Instrument (WPAI),Citation26 and a four-item assessment of satisfaction with treatment (treatment satisfaction questionnaire; TSQ).
Open-label extension study
The primary efficacy evaluation was the change from baseline in mean NRS pain score. The last on-therapy visit from the short-term study served as the baseline visit for the extension. Secondary efficacy endpoints were change from baseline in SIS, PGI-C, CGI-C, PGSR, and PGR scores.
Health outcomes assessments planned for the open-label extension trial were the EQ-5D, WPAI, and TSQ.
Safety assessments
Adverse events (AEs), early withdrawals due to AEs, and concomitant medications were recorded throughout both trials. Treatment-emergent adverse events (TEAEs) were defined as any AE that had its onset or worsened in severity during the on-therapy period. Safety assessments in both trials also included clinical laboratory determinations, vital sign measurements, standard 12-lead electrocardiogram (ECG) results, physical examinations, and Beck Depression Inventory-II and Michigan Neuropathy Screening Instrument scores.
Statistical methods
The sample size calculation for the short-term, placebo-controlled study was based on an analysis of covariance (ANCOVA) model on the primary efficacy variable, for the comparison between each dose level of desvenlafaxine (50, 100, 200, and 400 mg) and placebo. Approximately 90 patients per group was required for statistical significance at an error level of 1.25% (5% divided by 4) with a power of approximately 90%, using the Hochberg procedure to adjust for the multiplicity associated with testing four treatment groups against placebo. The sample size for the extension study was intended to obtain adequate long-term safety data in this patient population, hence was not powered formally.
For each study, the primary efficacy analysis set was the intent-to-treat (ITT) population, which included all patients who had a baseline efficacy evaluation, had received at least one dose of study medication, and had at least one primary efficacy evaluation during the on-treatment phase of the trial. The safety populations were composed of all patients who received at least one dose of double-blind or open-label study treatment in the short-term and extension studies, respectively.
Statistical analyses for both studies were conducted using SAS data analysis software (SAS Institute, Cary, NC, USA) and based on data from all sites. All tests associated with efficacy analyses were two-tailed and significance was based on alpha =0.05.
Short-term, double-blind study
The primary efficacy analysis was based on a mixed-effects model for repeated measures (MMRM) on the change from baseline in NRS score at week 13. The model included treatment, site, week, and interaction of treatment with week as factors and baseline value as the covariate. An unstructured covariance matrix was used to model the within-subject errors. Rate of response on the NRS was analyzed using logistic regression with treatment and site as factors and baseline mean pain score as a covariate. Response was defined as a reduction of 50% or more in weekly mean pain score from baseline to the endpoint (based on the mean of the last seven daily pain scores while on-therapy). Patients who withdrew due to AEs or lack of efficacy were considered as nonresponders in this analysis. Analysis of PGI-C and CGI-C used MMRM that included treatment, site, week, and interaction of treatment by week as factors. A similar MMRM was used to analyze changes from baseline in SIS, PGSR, and PGR scores; treatment, site, week, and interaction of treatment by week were included as factors, and the respective baseline value as the covariate. Changes from baseline in health outcomes scores were analyzed using ANCOVA, with treatment and site as factors and baseline score as the covariate; the last observation carried forward approach was used for handling missing data. The Hochberg adjustment for multiplicity was used in the primary analysis only. Safety data were summarized by dose and mean changes from baseline were analyzed using paired t-tests at the 5% significance level without adjustment for multiple tests.
A planned interim analysis was conducted on the initial 6 weeks of data from the first 225 randomly assigned patients to complete 6 weeks of treatment. The primary objectives of the interim analysis were to determine whether to stop ongoing randomization to desvenlafaxine doses in the current study, based on lack of efficacy or poor tolerability, and to inform the sponsor regarding Phase III study planning. An interim analysis board, composed of both sponsor and nonsponsor members, assessed the risk–benefit ratio for each desvenlafaxine dose using predefined decision rules.
Open-label extension study
Safety data were summarized and changes from baseline were evaluated for vital signs, weight, laboratory evaluations, and 12-lead ECGs using paired t-tests at the 5% significance level. The analyses of efficacy endpoints were descriptive in nature and only summarization of mean changes from baseline were reported. The last on-therapy evaluation from the short-term study served as the baseline for safety and efficacy assessments.
Results
Patient characteristics
Short-term, double-blind study
In the short-term study, 807 patients were screened and 412 were randomly assigned. Randomization to the 50 mg and 400 mg doses was halted after review of the interim analysis results, and all patients subsequently enrolled were randomized to receive either desvenlafaxine 100 mg/day, desvenlafaxine 200 mg/day, or placebo. The safety population included 408 patients, and 405 provided data for primary ITT efficacy analysis (). Demographic and baseline characteristics were generally balanced across the five treatment groups in the double-blind study (). The average age was 60.3 years. The study population was mostly male (73%) and white (87%). The mean (SD) NRS pain score at baseline was 6.45 (1.57). There was a statistically significant difference between groups in baseline weight; mean weight ranged from 100.6 kg to 111.0 kg across treatment groups (P=0.025; ). Baseline hemoglobin A1c levels ranged from 0.0713–0.0743 L/L for the treatment groups at baseline, and no between-groups differences were reported at week 13.
Figure 1 Study flowchart.
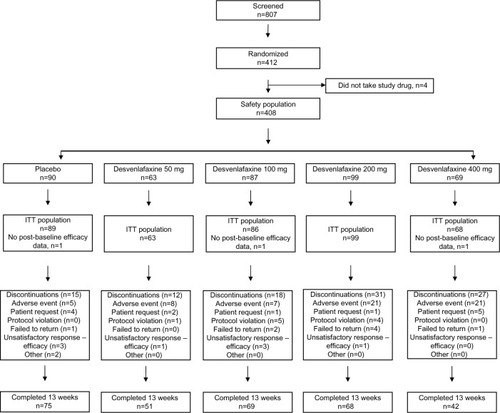
Table 1 Demographics and baseline characteristics, short-term study (safety population)
Open-label extension study
A total of 240 eligible patients from the double-blind study were enrolled in the open-label extension study (safety population: n=237; ITT population: n=223). The demographics for those individuals who enrolled in the extension study were similar to those in the overall safety population. The mean NRS at open-label baseline was 3.93 (SD: 2.15).
Efficacy
Short-term, double-blind study
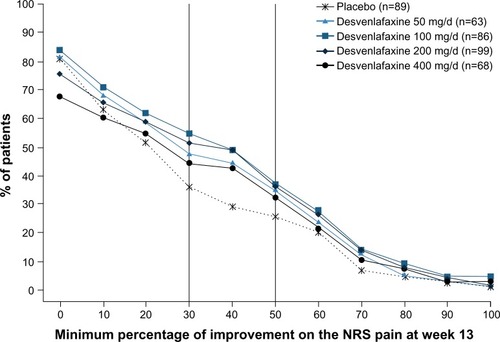
The NRS pain severity scores decreased with time in all treatment groups (). At week 13, the mean decrease in pain scores was significantly greater in the desvenlafaxine 200 mg (adjusted mean change, −2.93; difference from placebo, 1.10; adjusted Hochberg P=0.001) and 400 mg groups (adjusted mean change, −2.74; difference from placebo, 0.91; adjusted Hochberg P=0.027) compared with placebo (adjusted mean change: −1.83). The adjusted mean change in NRS score at week 13 for the 50 and 100 mg dose groups did not differ significantly from placebo (−2.41 and −2.42, respectively; both adjusted Hochberg P=0.084). At week 13, the proportion of patients with a 50% or greater reduction from baseline in NRS pain score (responders) was 26% for the placebo group and ranged from 32% to 37% for the desvenlafaxine dose groups (); differences from placebo did not reach statistical significance for any desvenlafaxine dose group. The proportions of patients in each treatment group achieving a range of thresholds of reduction in NRS score at week 13 are shown in .
Figure 2 Adjusted mean change from baseline on NRS pain severity over time (ITT; MMRM).
Abbreviations: ITT, intent-to-treat; MMRM, mixed-effects model for repeated measures; NRS, numeric rating scale.
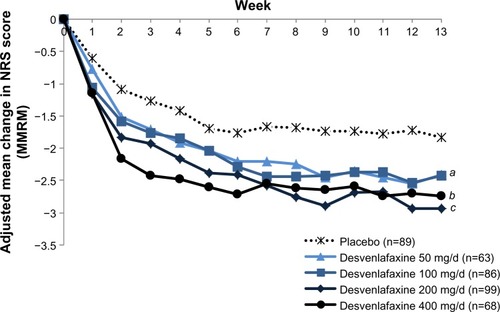
Figure 3 Response rate on pain severity score on the NRS at week 13 (ITT).
Abbreviations: ITT, intent-to-treat; NRS, numeric rating scale.
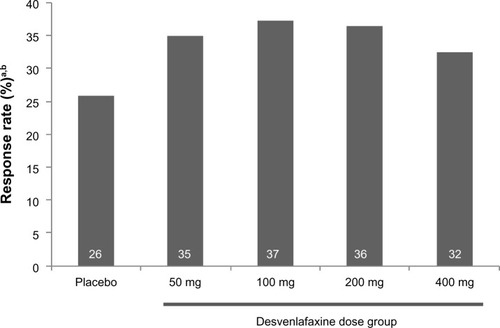
Figure 4 Proportions of patients achieving minimum thresholds of reduction in NRS score at week 13 (ITT).
Results of other secondary efficacy endpoints are summarized in . Statistically significant differences from placebo were found in PGI-C scores for all desvenlafaxine doses. Differences from placebo were significant for SIS scores at the desvenlafaxine 100, 200, and 400 mg doses (without adjustment for multiplicity).
Table 2 Secondary efficacy endpoints (MMRM), short-term study (intent-to-treat population)
Significant differences from placebo were observed at week 13 for some health outcomes measures at some desvenlafaxine doses. The adjusted mean change (standard error) from baseline in EQ-5D total score and TSQ overall score differed significantly from placebo for the desvenlafaxine 200 mg group only (EQ-5D: desvenlafaxine 200 mg, 0.15 [0.02]; placebo, 0.09 [0.02]; P=0.024; TSQ: desvenlafaxine 200 mg, 1.09 [0.14]; placebo, 0.62 [0.15]; P=0.025). Adjusted mean change in SF-36 physical component score was significantly different from placebo in the desvenlafaxine 50 mg dose group (desvenlafaxine 50 mg, 6.22 [0.90]; placebo, 3.50 [0.77]; P=0.022). No differences from placebo were observed for any desvenlafaxine dose group on the SF-36 mental component subscale scores or for POMS total mood disturbance score. The most consistent improvement compared with placebo was observed for WPAI-activity impairment (WPAI-AI) scores: the adjusted mean change from baseline was significantly different from placebo (−0.10 [0.03]) for all four desvenlafaxine dose groups, ranging from −0.18 (0.03) for desvenlafaxine 50 mg/day and 200 mg/day (P=0.048 and P=0.017, respectively) to −0.22 (0.03; P=0.002) for desvenlafaxine 400 mg/day. For the WPAI-work impairment (WPAI-WI) score, separation from placebo was only seen with the 100 mg dose (−0.18 [0.03]; placebo, –0.09 [0.03]; P=0.043). Health outcomes analyses were not controlled for multiplicity.
Open-label extension study
A total of 147 patients were exposed to a mean daily desvenlafaxine dose of 238 mg for at least 240 days. Mean daily desvenlafaxine dose ranged from 228.2 mg/day to 254.6 mg/day between study visits from week 3 (after the first 2 weeks at 100 mg/day) to month 9.
The open-label extension study was prematurely terminated for business reasons. The primary efficacy endpoint was summarized descriptively and no formal statistical analyses were performed. There was a decrease from open-label baseline in mean NRS scores at month 9 of −0.53. The mean NRS pain score decreased from a baseline of 3.86 to 3.35 at the final on-therapy evaluation. No formal statistical analyses were conducted for secondary efficacy and health outcomes endpoints due to early termination of the study.
Safety and tolerability
Short-term, double-blind study
Mean daily doses at study week 13 (including days missed) were 49.7 mg/day, 99.6 mg/day, 199.1 mg/day, and 392.6 mg/day for the desvenlafaxine 50, 100, 200, and 400 mg groups, respectively. No dosage reductions due to tolerability were reported.
The most common TEAEs (reported by ≥5% patients in any desvenlafaxine group and at twice the placebo rate) were nausea and dizziness, with TEAEs occurring in a dose-dependent manner (). Taper/post-therapy emergent AEs reported by more than one patient in any treatment group were nausea, vomiting, and dizziness, which were all reported at desvenlafaxine doses of 100 mg/day or higher. A total of 62/408 (15%) patients discontinued the short-term study due to AEs; nausea and dizziness were the AEs most commonly cited as the reason for discontinuation. Rates of discontinuations due to AEs were dose-dependent: five (5.6%) patients discontinued due to AEs in the placebo group compared with eight (12.7%) in the desvenlafaxine 50 mg group, seven (8%) in the desvenlafaxine 100 mg group, 21 (21.2%) in the desvenlafaxine 200 mg group, and 21 (30.4%) in the desvenlafaxine 400 mg group. The majority of discontinuations due to AEs (32 of the total 62) occurred during the first 2 weeks of treatment.
Table 3 Treatment-emergent adverse events in ≥5% and twice that of placebo for patients in any treatment group, short-term study (safety population)
A total of 12 patients reported serious adverse events (SAEs); six patients in the placebo group, one each in the desvenlafaxine 50, 100, and 200 mg groups, and three (one in the prestudy period) in the desvenlafaxine 400 mg group. Cardiac SAEs included one report of angina pectoris (placebo) and one report of ventricular tachycardia (desvenlafaxine 200 mg/day). One overdose was reported by a patient receiving placebo. One SAE (thyroid cancer) reported in the 400 mg group resulted in death after the patient had withdrawn from the study. Other SAEs included pneumonia, two fractures, and an increase in blood glucose (574 mg/dL; resolved with intravenous fluids and insulin treatment) in placebo-treated patients, and pneumonia, gastrointestinal hemorrhage, esophageal cancer, colon cancer, mental status changes (after hemicolectomy with ileocolonic anastomosis), urinary retention, and orthostatic hypotension in desvenlafaxine-treated patients.
Laboratory evaluations with significant changes from baseline compared with placebo at week 13 are reported in . Significant increases in alkaline phosphatase, total cholesterol, and high density lipoprotein cholesterol were observed at week 13 for one or more desvenlafaxine group, and for each, changes from baseline were dose-related. Small mean increases from baseline in supine heart rate and diastolic blood pressure (BP) were observed at various time points in each dose group, but there were no significant differences compared with placebo at week 13. Changes from baseline at week 13 in weight and 12-lead heart rate are shown in . Mean weight decreased in a dose-dependent manner, and adjusted mean decreases in weight at week 13 were statistically significant compared with placebo for the desvenlafaxine 100, 200, and 400 mg doses. Two patients, both treated with desvenlafaxine 400 mg/day, had clinically important weight losses of 10.3 and 12.7 kg, respectively.
Table 4 Selected vital signs and laboratory values at week 13 (LOCF), short-term study (safety population)
Open-label extension
During the 9-month extension, 147 patients were exposed to a mean daily desvenlafaxine dose of 238 mg for at least 240 days. Overall, the safety and tolerability profile of long-term desvenlafaxine treatment was similar to that observed in the short-term study. During the on-therapy period, 80.2% of patients reported TEAEs, the most common of which (reported by >5%) were dizziness (12.2%), nausea (8.4%), upper respiratory tract infection (8.4%), hypertension (8.0%), nasopharyngitis (7.2%), dry mouth (6.3%), arthralgia (5.5%), headache (5.5%), vomiting (5.5%), fatigue (5.1%), and back pain (5.1%). Dizziness (6.3%) and headache (2.5%) were the most frequently reported AEs during the taper and post-study period. A total of 37 (15.6%) patients discontinued study treatment due to AEs across the 9-month study period. The most common AEs that led to discontinuation were dizziness (2.1%) and nausea (1.7%).
Serious adverse events were reported by 26 (11%) patients. Chest pain and congestive heart failure each occurred in three patients and headache was reported in two patients. SAEs occurring in three patients were considered related to study treatment: chest pain, erosive esophagitis, and hiccup in one patient, increased BP in one patient, and mental status changes in the third. All other SAEs were considered unrelated to study treatment. No deaths occurred during the open-label extension study.
At the 9-month evaluation, there were increases in mean glycosylated hemoglobin Alc (+0.0023 L/L), serum creatinine (+2.3 μmol/L), total cholesterol (+0.16 mmol/L), and triglycerides (+0.168 mmol/L). Decreases were observed in mean total bilirubin levels (−0.96 μmol/L), hemoglobin (−2.78 g/L), and hematocrit (−0.00439 L/L). A total of 56 patients had laboratory results of clinical importance: abnormal liver function test (one patient), decreased hematocrit (1), decreased hemoglobin (1), glucose levels (21), increased triglycerides (4), increased cholesterol (1), and urine protein levels (27). Mean weight and pulse did not differ significantly from baseline at 9 months; supine diastolic BP increased 1.39 mmHg. Two patients had clinically important weight changes; one lost 10.4 kg (desvenlafaxine 100 mg/day), and the second gained 9.5 kg (desvenlafaxine 200 mg/day). Clinically important vital signs results included postural hypotension in four patients, clinically important changes in systolic BP in 32 patients, and clinically important changes in diastolic BP in 31 patients. Four patients exhibited changes on ECG that were considered clinically important.
Discussion
The short-term, double-blind study of desvenlafaxine treatment in patients with DPN is the first clinical trial to demonstrate the analgesic effect of desvenlafaxine in a chronic pain condition. In the short-term trial, there was a dose-dependent effect of desvenlafaxine on both efficacy and tolerability. Efficacy measures showed significance at the higher dose groups (200 mg and 400 mg). Differences from placebo on NRS score for the two desvenlafaxine doses − 1.10 and 0.91, respectively – were similar to effect sizes reported for duloxetine (−1.13 [24-hour pain score]) and pregabalin (−0.90 [24-hour pain score]) in a meta-analysis of eleven DPN trials (including three duloxetine and six pregabalin trials).Citation9 The change in NRS score for the 200 and 400 mg doses (adjusted mean change, −2.93 and −2.74, respectively) also exceeded the two-point change found to be associated with clinically important improvement on the NRS scaleCitation21 whereas the change from baseline for placebo did not (adjusted mean change, −1.83). However, the efficacy benefit of the higher desvenlafaxine doses was mitigated by a clear increase in the incidence of AEs and study withdrawal due to AEs, particularly at the 400 mg dose. Although desvenlafaxine was safe and generally well-tolerated at all doses evaluated, the higher doses needed for pain efficacy did not achieve the positive tolerability profile observed for the 50 mg/day doseCitation27 recommended for the treatment of MDD;Citation19 the development program for the treatment of pain was discontinued.
Patients in all desvenlafaxine dose groups showed significant improvement compared with placebo based on PGI-C scores. Results for other secondary efficacy endpoints were not consistent across doses (): one to three dose groups showed significant change for each secondary measure, with no apparent dose-related trend across measures. For the PGSR, only the highest dose differed significantly from placebo, whereas only the 50 mg dose had statistically significant improvement versus placebo on the PGR. Desvenlafaxine treatment was also associated with improvement in functional outcomes. Patients in all four desvenlafaxine dose groups showed significant improvement in WPAI activity impairment scores compared with placebo. To our knowledge, this is the first report to demonstrate improvement on the WPAI-AI, and the finding suggests that pain improvement with desvenlafaxine treatment was accompanied by greater functional activity. Significant improvement on the WPAI-WI scale was reported for desvenlafaxine 100 mg versus placebo. The lack of significant improvement on this scale at higher doses may be related to the higher rates of AEs reported at these doses. Significant improvement in EQ-5D total score, which measures mobility, self-care, usual activities, pain/discomfort, and anxiety/depression, was observed in the desvenlafaxine 200 mg/day group only. The EQ-5D has been included in several published studies of duloxetine or pregabalin for DPN, and significant improvement has been reported in some of these studies,Citation28–Citation31 but not others.Citation32–Citation34
The current study extends the set of randomized clinical trials examining the efficacy of serotonergic/noradrenergic antidepressant drugs for treating pain associated with DPN. Duloxetine is the most extensively studied of the SNRIs and has demonstrated efficacy versus placebo in several short-term clinical trials,Citation28,Citation30,Citation35,Citation36 and positive results in placebo-controlled trials have been reported for the SNRI venlafaxine.Citation20,Citation37 TCAs, including amitriptyline, desipramine, and clomipramine, have also demonstrated statistically significant improvement in pain associated with DPN compared with placebo.Citation38–Citation40 Drugs from the two classes of antidepressants appear to be similar in efficacy to anticonvulsant drugs, based on both head-to-headCitation35 and indirect comparisons,Citation41 although a network meta-analysis found gabapentin to have a more favorable balance between efficacy and tolerability compared with duloxetine, venlafaxine, and amitriptyline.Citation41 The efficacy and tolerability results from the current study are consistent with the findings of that analysis.
The short-term study used an adaptive design in which an interim analysis was performed approximately halfway through the study. The adaptive design allowed for initial assessment of a broad range of doses. Discontinuation of randomization was planned for those doses found to be either ineffective or poorly tolerated, with all patients subsequently randomly assigned to the remaining doses. Following the interim analysis in the short-term trial, randomization to the 50 mg and 400 mg doses of desvenlafaxine was discontinued; randomization to the 100 mg and 200 mg doses continued. The long-term extension study was terminated before completion for business reasons. However, safety and tolerability findings from the 9-month trial contribute to the overall safety profile of desvenlafaxine. The most common AEs reported in the short-term and long-term studies (nausea, dizziness), and the observed dose-related increases in AEs and discontinuations due to AEs are consistent with the tolerability profile for desvenlafaxine observed in MDD patients.Citation27 No new or unexpected safety signals were observed in this population of DPN patients.
Several factors limit the generalizability of the results reported. First, only patients who were medically stable were eligible for enrollment. Based on the study inclusion criteria, patients required written documentation of stable and optimized glycemic control for at least 3 months before randomization. The exclusion is standard for DPN studies and therefore does not affect the generalizability of this study relative to other clinical trials. However, the written requirement substantially reduced the number of eligible patients, and limits generalization of the findings to a stabilized diabetic patient population. Patients with other clinically significant disease were also excluded, limiting the generalizability to patients with conditions commonly associated with diabetes mellitus, such as hypertension, coronary heart disease, and stroke.Citation42 In addition, the short-term, double-blind trial used fixed desvenlafaxine doses. Some patients may not have received a dose sufficient for symptom improvement, whereas others may have received a higher dose than needed, and consequently experienced poorer tolerability. Finally, the collection of AEs by spontaneous report may have underestimated rates of AEs compared with the use of a quantitative assessment.
Conclusion
Desvenlafaxine treatment was effective in reducing pain in patients with DPN, at doses (≥200 mg/day) higher than the 50 mg dose recommended for the treatment of MDD. Overall, the study showed desvenlafaxine to be generally well-tolerated in this study population. There was, however, a dose-dependent increase in the incidence of AEs and withdrawals due to AEs.
Acknowledgments/disclosure
This study was sponsored by Wyeth, which was acquired by Pfizer in October 2009. Medical writing support for this manuscript was provided by Kathleen M Dorries, PhD, of Peloton Advantage and was funded by Pfizer.
Rob Allen, is a former Pfizer employee currently working as an independent consultant. Suna Barlas, is a Pfizer employee. Uma Sharma, is a former Pfizer employee and currently works at MMS Holdings Inc.
References
- TesfayeSVileikyteLRaymanGToronto Expert Panel on Diabetic NeuropathyPainful diabetic peripheral neuropathy: consensus recommendations on diagnosis, assessment and managementDiabetes Metab Res Rev201127629638
- JensenTSBackonjaMMHernandezJSTesfayeSValensiPZieglerDNew perspectives on the management of diabetic peripheral neuropathic painDiab Vasc Dis Res2006310811917058631
- WongMCChungJWWongTKEffects of treatments for symptoms of painful diabetic neuropathy: systematic reviewBMJ20073358717562735
- LindsayTJRodgersBCSavathVHettingerKTreating diabetic peripheral neuropathic painAm Fam Physician20108215115820642268
- AttalNCruccuGHaanpääMEFNS Task ForceEFNS guidelines on pharmacological treatment of neuropathic painEur J Neurol2006131153116917038030
- BenyaminRTrescotAMDattaSOpioid complications and side effectsPain Physician200811S105S12018443635
- Lyrica [package insert]New York, NYPfizer, Inc2012
- Cymbalta [package insert]Indianapolis, INEli Lilly and Company2011
- QuiliciSChancellorJLöthgrenMMeta-analysis of duloxetine vs pregabalin and gabapentin in the treatment of diabetic peripheral neuropathic painBMC Neurol20099619208243
- KaurHHotaDBhansaliADuttaPBansalDChakrabartiAA comparative evaluation of amitriptyline and duloxetine in painful diabetic neuropathy: a randomized, double-blind, cross-over clinical trialDiabetes Care20113481882221355098
- BansalDBhansaliAHotaDChakrabartiADuttaPAmitriptyline vs pregabalin in painful diabetic neuropathy: a randomized double blind clinical trialDiabet Med2009261019102619900234
- BrilVEnglandJFranklinGMAmerican Academy of NeurologyAmerican Association of Neuromuscular and Electrodiagnostic MedicineAmerican Academy of Physical Medicine and RehabilitationEvidence-based guideline: Treatment of painful diabetic neuropathy: report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and RehabilitationNeurology2011761758176521482920
- SelvarajahDWilkinsonIDDaviesJGandhiRTesfayeSCentral nervous system involvement in diabetic neuropathyCurr Diab Rep20111131032221667355
- MarksDMShahMJPatkarAAMasandPSParkGYPaeCUSerotonin-norepinephrine reuptake inhibitors for pain control: premise and promiseCurr Neuropharmacol2009733133620514212
- DeecherDCBeyerCEJohnstonGDesvenlafaxine succinate: A new serotonin and norepinephrine reuptake inhibitorJ Pharmacol Exp Ther200631865766516675639
- BoyerPMontgomerySLepolaUEfficacy, safety, and tolerability of fixed-dose desvenlafaxine 50 and 100 mg/day for major depressive disorder in a placebo-controlled trialInt Clin Psychopharmacol20082324325318703933
- LiebowitzMRManleyALPadmanabhanSKGangulyRTummalaRTourianKAEfficacy, safety, and tolerability of desvenlafaxine 50 mg/day and 100 mg/day in outpatients with major depressive disorderCurr Med Res Opin2008241877189018507895
- DunlopBWReddySYangLLubaczewskiSFochtKGuico-PabiaCJSymptomatic and functional improvement in employed depressed patients: a double-blind clinical trial of desvenlafaxine versus placeboJ Clin Psychopharmacol20113156957621869698
- Pristiq [package insert]Philadelphia, PAWyeth Pharmaceuticals, Inc, a subsidiary of Pfizer Inc2013
- RowbothamMCGoliVKunzNRLeiDVenlafaxine extended release in the treatment of painful diabetic neuropathy: a double-blind, placebo-controlled studyPain200411069770615288411
- FarrarJTYoungJPJrLamoreauxLWerthJLPooleRMClinical importance of changes in chronic pain intensity measured on an 11-point numerical pain rating scalePain20019414915811690728
- FeldmanELStevensMJClinical testing in diabetic peripheral neuropathyCan J Neurol Sci1994214S3S77874610
- WareJEJrSherbourneCDThe MOS 36-item short-form health survey (SF-36)Medical Care1992304734831593914
- LorrMMcNairDMDropplemanLFProfile of mood states technical updateMulti-Health Systems, Inc2005
- EuroQol GroupEuroQol – a new facility for the measurement of health-related quality of lifeHealth Policy19901619920810109801
- ReillyMCZbrozekASDukesEMThe validity and reproducibility of a work productivity and activity impairment instrumentPharmacoeconomics1993435336510146874
- ClaytonAHKornsteinSGRosasGGuico-PabiaCTourianKAAn integrated analysis of the safety and tolerability of desvenlafaxine compared with placebo in the treatment of major depressive disorderCNS Spectr20091418319519407730
- WernickeJFPritchettYLD’SouzaDNA randomized controlled trial of duloxetine in diabetic peripheral neuropathic painNeurology2006671411142017060567
- ArmstrongDGChappellASLeTKDuloxetine for the management of diabetic peripheral neuropathic pain: evaluation of functional outcomesPain Med2007841041817661854
- GoldsteinDJLuYDetkeMJLeeTCIyengarSDuloxetine vs placebo in patients with painful diabetic neuropathyPain200511610911815927394
- TolleTFreynhagenRVersavelMTrostmannUYoungJPJrPregabalin for relief of neuropathic pain associated with diabetic neuropathy: a randomized, double-blind studyEur J Pain20081220321317631400
- GaoYNingGJiaWPDuloxetine versus placebo in the treatment of patients with diabetic neuropathic pain in ChinaChin Med J (Engl)20101233184319221163113
- MoonDELeeDILeeSCEfficacy and tolerability of pregabalin using a flexible, optimized dose schedule in Korean patients with peripheral neuropathic pain: a 10-week, randomized, double-blind, placebo-controlled, multicenter studyClin Ther2010322370238521353106
- GilronIWajsbrotDTherrienFLemayJPregabalin for peripheral neuropathic pain: a multicenter, enriched enrollment randomized withdrawal placebo-controlled trialClin J Pain20112718519321178603
- BoyleJErikssonMEGribbleLRandomized, placebo-controlled comparison of amitriptyline, duloxetine, and pregabalin in patients with chronic diabetic peripheral neuropathic pain: impact on pain, polysomnographic sleep, daytime functioning, and quality of lifeDiabetes Care2012352451245822991449
- RaskinJPritchettYLWangFA double-blind, randomized multicenter trial comparing duloxetine with placebo in the management of diabetic peripheral neuropathic painPain Med2005634635616266355
- KadirogluAKSitDKayabasiHTuzcuAKTasdemirNYilmazMEThe effect of venlafaxine HCl on painful peripheral diabetic neuropathy in patients with type 2 diabetes mellitusJ Diabetes Complications20082224124518413214
- MaxMBKishore-KumarRSchaferSCEfficacy of desipramine in painful diabetic neuropathy: a placebo-controlled trialPain199145391861872
- MaxMBLynchSAMuirJShoafSESmollerBDubnerREffects of desipramine, amitriptyline, and fluoxetine on pain in diabetic neuropathyN Engl J Med1992326125012561560801
- SindrupSHGramLFSkjoldTGrodumEBrosenKBeck-NielsenHClomipramine vs desipramine vs placebo in the treatment of diabetic neuropathy symptoms. A double-blind cross-over studyBr J Clin Pharmacol1990306836912271367
- RudrojuNBansalDTalakokkulaSTComparative efficacy and safety of six antidepressants and anticonvulsants in painful diabetic neuropathy: a network meta-analysisPain Physician201316E705E71424284851
- KalyaniRRSaudekCDBrancatiFLSelvinEAssociation of diabetes, comorbidities, and A1c with functional disability in older adults: results from the National Health and Nutrition Examination Survey (NHANES), 1999–2006Diabetes Care2010331055106020185736