
Abstract
Non-small-cell lung cancer (NSCLC) patients with mutated or rearranged oncogene drivers can be treated with upfront selective inhibitors achieving higher response rates and longer survival than chemotherapy. The RET gene can undergo chromosomal rearrangements in 1%–2% of all NSCLC patients, involving various upstream fusion partners such as KIF5B, CCDC6, NCOA4, and TRIM33. Many multikinase inhibitors are active against rearranged RET. Cabozantinib, vandetanib, sunitinib, lenvatinib, and nintedanib achieved tumor responses in about 30% of these patients in retrospective studies. Prospective phase II trials investigated the activity and toxicity of cabozantinib, vandetanib, sorafenib, and lenvatinib, and did not reach significantly higher response rates. VEGFR and EGFR inhibition represented the main ways of developing off-target toxicity. An intrinsic resistance emerged according to the type of RET fusion partners, as KIF5B-RET fusion is the most resistant. Also acquired mutations in rearranged RET oncogene developed as resistance to these multikinase inhibitors. Interestingly, RET fusions have been found as a resistance mechanism to EGFR-TKIs in EGFR-mutant NSCLC patients. The combination of EGFR and RET inhibition can overcome this resistance. The limitations in terms of activity and tolerability of the various multikinase inhibitors prompted the investigation of new highly selective RET inhibitors, such as RXDX-105, BLU-667, and LOXO-292. Some data emerged about intracranial antitumor activity of BLU-667 and LOXO-292. If these novel drugs will achieve high activity in RET rearranged NSCLC, also these oncogene-addicted tumors can undergo a significant survival improvement.
Introduction
Oncogene addiction is a phenomenon identified in many neoplasms. It is relevant for both carcinogenesis and cancer progression. Recently, targeting oncogene drivers has become one of the main cancer treatment strategies. We can mention some examples, such as Abelson tyrosin-protein kinase 1 (ABL1) inhibitors in chronic myeloid leukemia, epidermal growth factor receptor (EGFR), anaplastic lymphoma kinase (ALK), and c-ros proto-oncogene (ROS1) inhibitors in non-small-cell lung cancer (NSCLC); BRAF and MEK inhibitors in melanoma and NSCLC. Mutations or chromosomal rearrangement involving these oncogenes can be easily detected in tumor samples to guide decision-making of optimal cancer treatment.
In the upfront management of advanced NSCLC, the classification in oncogene-addicted and non-oncogene-addicted tumors is crucial to address patients to the proper treatment: first-line chemotherapy and/or immunotherapy if their tumor is non-oncogene-addicted and kinase inhibitors if oncogene addiction is documented via specific gene alterations (eg, EGFR mutations, ALK or ROS1 rearrangements, BRAF mutations).Citation1,Citation2
The identification of mutated or rearranged oncogene drivers and consequent first-line treatment of NSCLC patients with selective inhibitors achieved high response rates, not usually achievable through chemotherapy. These outcomes in terms of tumor response can be translated in longer survival with an improvement of quality-of-life and manageable side-effects.
In all these cases, even in those with longer progression-free survival, resistance occurs. To date many resistance mechanisms have been known and, as a consequence, novel targeted drugs have been developed to deal with the inefficacy of previous treatment. Resistance mutations can be detected through the re-biopsy of tumor tissue or the so-called “liquid biopsy”, intended as the analysis of blood samples to find DNA alterations.Citation3 Among the various oncogene drivers in NSCLC the RET gene is involved in various chromosomal rearrangements, which can be found in 1%–2% of all NSCLC patients.Citation4 The current availability of RET inhibitors raises the possibility to spare this small proportion of NSCLC patients from chemotherapy and offer the opportunity of a further effective targeted therapy after failure of chemotherapy.
In this review, we discuss the biological significance of the RET gene, the available RET-directed drugs, and relative clinical trials for NSCLC patients and resistance mechanisms occurring during the treatment with RET inhibitors.
RET function and its alterations in carcinogenesis
In 1985, RET was identified as a novel transforming gene as result of transfection of the NIH 3T3 cell with high molecular weight DNA of a human T-cell lymphoma. The gene was activated by a DNA rearrangement in which two unlinked segments of human DNA recombined to generate a new transcriptional unit.Citation5 Subsequently, studies mapped RET to chromosome 10q11.2, where it encodes for a receptor tyrosine kinase.Citation6
Mutations of RET determine the absence of enteric ganglia from the distal colon (Hirschsprung’s disease) and congenital megacolon, besides RET.k-/RET.k- mice lack all enteric ganglia, demonstrating a RET important role in the development of the enteric nervous system.Citation7 Still in the embryonic phase a RET-dependent cell rearrangement generates a specialized epithelial domain that later emerges as the tip of the primary ureteric bud.Citation8 As well as in the embryonic phase RET is important in homeostasis of several tissues including neural, neuroendocrine, hematopoietic, and male germ tissues.Citation9
RET is a single-pass transmembrane protein with a typical intracellular tyrosine kinase domain (). While a “classical” activation of a receptor tyrosine kinase (RTK) is due to the interaction ligand-receptor, the activation of RET requires an interaction between its ligands (the glial cell line-derived neurotrophic factor-family ligands, GFLs) and a co-receptor (GFLs family receptor-alfa).Citation5 The GFL–GFRα complex binds to the extracellular domain of RET, leading to the phosphorylation of the intracellular tyrosine kinase domain and consequently the activation of several pathways, including MAPK, PI3K, JAK-STAT, PKA, and PKC.Citation10
Figure 1 Schematic structure of wild-type and rearranged RET proteins in a cancer cell.
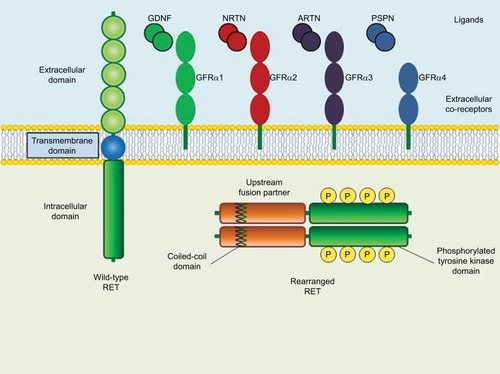
Multiple endocrine neoplasia (MEN) syndrome is defined as a disorder with neoplasms in two or more different hormonal tissues. Each of these syndromes also includes one or more additional neoplasms. MEN1 is characterized by specific hormonal tumors: parathyroid adenoma (90%), gastrinoma (40%), and prolactinoma (30%), plus additional hormone-producing tumor with a range between 1% and 10% (insulinoma, glucagonoma, VIPoma, somatostatinoma, pituitary tumors, thymic carcinoid, bronchial carcinoid, gastric carcinoid, adrenal cortex, and pheochromocytoma).Citation11 The MEN1 gene, mapped at chromosome 11q13, encodes for a protein, menin, that is involved in organogenesis of neural tube, heart, and craniofacial structures and hematopoiesis.Citation12 A germline mutation of the MEN1 leads to tumor development mainly via a biallelic loss-of-function mechanism.Citation11 MEN2A are characterized by medullary thyroid cancer, pheochromocytoma, and hyperparathyroidism. MEN2B has the same features as MEN2A, plus intestinal ganglioneuromas and the mucosal neuroma phenotype, but a different hormonal disorder profile.Citation13 During the 90 seconds, the International RET Mutation Consortium enrolled 477 independent MEN2 families worldwide to investigate the association between the position and type of germline mutation in the RET proto-oncogene and the presence or absence of medullary thyroid carcinoma, pheochromocytoma, primary hyperparathyroidism, and other. Overall, 92% of MEN2 families had a germline RET mutation in one of eight codons. Over 95% of families with MEN2B had a mutation in codon 918, and each of the three MEN2A categories were found to have mutations at cysteine codons 609, 611, 618, 620, and 634.Citation14 These mutations determine a ligand-independent constitutive activation of the tyrosine kinase receptor leading to an uncontrolled activation of the MAPK and the PI3K pathways that results in uncontrolled growth and cell de-differentiation.Citation15 In addition to the RET point mutations, several gene rearrangements were identified in papillary thyroid carcinoma (PTC), known as RET/PTC rearrangements. Each distinct chromosomal translocation is characterized by the promoter and 5′ region of a heterologous gene encoding a thyrocyte-expressed protein fused, in frame, to the kinase-encoding 3′ end of the RET proto-oncogene.Citation16 To date, 13 different oncogenic RET/PTC fusion proteins (termed RET/PTC1-PTC9) have been discovered. These chimeric proteins are characterized by coiled-coil domains that induce dimerization and activation of the kinase domain. This capability to form dimers is required for oncogenic activation, leading to an uncontrolled activation of the MAPK and PI3K pathways, similarly to the result of activating RET point mutations.Citation15,Citation17 Interestingly, RET rearrangement are largely thought to be somatic events, as opposed to RET mutations that can occur in the germ line or be acquired somatically.
First, in 2011 RET rearrangement was discovered in a young never-smoking male patient with lung adenocarcinoma.Citation18 As aforementioned, 1%–2% of NSCLCs harbor a RET rearrangement.Citation4 While the correlation between ionic radiation and RET rearrangements is clearly confirmed in PTC, in NSCLC this correlation remains unclear, even if in vitro experiments demonstrated the possibility to induce RET rearrangement in human lung cancer cells by radiation.Citation19 To deepen the knowledge of RET in NSCLC, Wang et alCitation20 examined the RET fusion gene in 936 patients with surgically resected NSCLC using a PCR strategy. These patients seemed to have identifiable clinicopathologic characteristics, including younger age, never-smoker status, early lymph node metastases, poor differentiation, and a solid-predominant subtype. As well as the other main driving-mutations, RET rearrangement seems to be mutually exclusive, suggesting that it might be a targetable oncogenic driver.Citation21
The first RET rearrangement identified in NSCLC patients was an in-frame fusion transcript of the kinesin family 5B gene (KIF5B) with RET gene (KIF5B-RET). Other upstream fusion partners for RET rearrangement have been identified in NSCLC, such as the coiled-coil domain-containing protein 6 (CCDC6), the nuclear receptor coactivator 4 (NCOA4), the tripartite motif-containing 33 (TRIM33), myosin VC gene (MYO5C), EPH receptor A5 gene (EPHA5), CAP-Gly domain containing linker protein family member one gene (CLIP1), ELKS/RAB6-interacting/CAST family member one gene (ERC1), phosphatidylinositol binding clathrin assembly protein gene (PICALM), FERM domain containing 4A gene (FRMD4A) RUN and RYVE domain containing two gene (RUFY2), and tripartite motif containing 24 gene (TRIM24). All of these fusion counterparts have a dimerization domain that induces ligand-independent activation of the RET kinase ().Citation22,Citation23
RET-directed drugs in lung cancer
The majority of drugs active against RET are multikinase inhibitors. The approval of these agents is not restricted to patients harboring alterations in RET gene. Among these drugs, we can mention those approved for thyroid cancer, such as cabozantinib, vandetanib, lenvatinib, and sorafenib, but also other multikinase inhibitors approved for other malignancies, including alectinib, sunitinib, nintedanib, regorafenib, and ponatinib. The action of these drugs against RET kinase allows a classification in type I and type II inhibitors. The first ones (ie, vandetanib and sunitinib) bind the ATP-binding domain in its active conformation. The latter ones bind the same domain, but in the inactive conformation.Citation24,Citation25
The effects of these drugs on RET rearrangements with various upstream partners or breakpoints were studied by means of engineered and patient-derived RET-rearranged cell lines and xenograft models.Citation26 These experimental models have been useful to identify the activity of other agents against RET rearranged kinase (eg, RXDX-105, BLU-667, LOXO-292).Citation27–Citation30 These studies demonstrated the effect on rearranged RET kinase, but the multikinase inhibition also induced the blockade of many downstream pathways such as MAPK, PI3K, and PLCγ with a consequent decrease of cell proliferation.
In 2015, a retrospective analysis was performed on data from the Global Multicenter RET Registry (GLORY), which collected the experiences of the treatment with multikinase inhibitors in RET-rearranged NSCLC patients.Citation31 Interesting findings emerge from this analysis. Multikinase RET inhibitors were administered in various lines of systemic therapy ranging from first to eighth, with a median of third line. In fact, median time from diagnosis to the beginning of anti-RET therapy was 12 months. Among the various drugs only cabozantinib, vandetanib, sunitinib, lenvatinib, and nintedanib achieved tumor responses, ~30%, whereas no responses were observed with alectinib, regorafenib, sorafenib, or ponatinib (). None of the outcome measures (response rate, progression-free survival [PFS], overall survival [OS]) changed depending on upstream fusion partners (eg, KIF5B, CCDC6, EPHA5) of RET gene. Median PFS of 2.3 months and median OS of 6.8 months were reported. The majority of patients (about 80%) received only one multikinase RET inhibitor. Moreover, this registry also provides information about the efficacy of first-line platinum-based chemotherapy in RET-rearranged NSCLC, which reached about 50%. These results in terms of response rate and PFS are partially concordant with those from phase II trials, which had studied or were studying the same drugs in RET-rearranged NSCLC patients.
Figure 2 (A) Response rates of retrospective analysis on anti-RET multikinase inhibitors from GLORY. (B) Response rates of 5 phase II trials on anti-RET multikinase inhibitors.
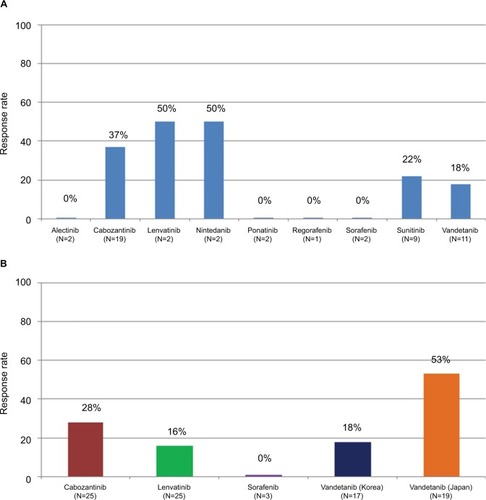
To date in this subpopulation, five phase II trials with multikinase RET inhibitors have been completed (). One single arm phase II trial studied cabozantinib, a multikinase inhibitor active against VEGFR2, MET, ROS1, AXL, KIT, and TIE2, but with low activity against RET (IC50=5.2 nM).Citation32 The patients in this study were not previously treated with RET inhibitors. About one-third of these patients responded to cabozantinib, but no complete responses were observed. Moreover, responses were early, with a high tumor shrinkage (≥30% tumor reduction in 70% of patients). The median overall survival reached 9.9 months.
Vandetanib is another multikinase inhibitor against VEG-FRs, EGFR, and RET, with higher IC50 than cabozantinib. It was studied in two phase II trials conducted in Eastern countries (Korea and Japan).Citation33,Citation34 The objective response rate (ORR) was 18% and 53%, for median PFS of 4.5 and 4.7 months, respectively. The most common adverse effects of vandetanib were hypertension because of VEGFR inhibition, skin toxicity and diarrhea because of EGFR inhibition, and also manageable prolonged QT interval. Interestingly in these studies the differences in the types of upstream fusion partners of RET gene were documented. In the Korean study, the KIF5B-RET rearrangement was associated with no objective response, unlike CCDC6-RET fusion. They also found the novel rearrangement MYO5C-RET, which is characterized by the exclusion of RET transmembrane domain with consequent ligand-independent RET activation. Accordingly in the Japanese trial higher ORR (83% vs 20%) and longer PFS (8.3 vs 2.9 months) were achieved with CCDC6-RET than KIF5B-RET rearrangements.
Also sorafenib was studied in a phase II trial for NSCLC patients with RET rearrangement.Citation35 Sorafenib is a multikinase inhibitor which targets intracellular (ie, CRAF, BRAF and mutated BRAF) and cell surface (ie, KIT, FLT3, VEGFRs and RET) molecules. It has anti-RET activity with IC50=5.9–47 nM. In a preclinical model, sorafenib was active against KIF5B-RET fusion.Citation36 In the only three patients treated with sorafenib in this study, no significant response was reported, but tumor shrinkage and symptom improvement were observed along with durable stable disease in one patient.
Finally, lenvatinib, a multikinase inhibitor against VEGFRs, PDGFR-β, and RET, has an IC50 for anti-RET activity of 35 nM. It was evaluated for antitumor activity in RET fusion positive patients with lung adenocarcinoma within a phase II study. Among 25 patients, ORR was 16% and median PFS 7.3 months. Grade 3–4 adverse events were experienced in 92% of patients, with hypertension, nausea, anorexia, diarrhea, proteinuria, and vomiting as the most common toxicities.Citation37
Resistance to RET inhibition
The results of these clinical trials with multikinase inhibitors have revealed that not all RET-rearranged patients are responsive to these drugs. Objective response rates of 28% and 47%,Citation32,Citation34 respectively obtained with cabozantinib and vandetanib, suggest the existence of intrinsic resistance mechanisms. First of all, the type of RET fusion partner seems to influence the response to treatment, as documented with vandetanib, which induced worse outcomes in the presence of KIF5B-RET fusion than with CCDC6-RET fusion. However, the phase II study with cabozantinib did not confirm this finding. Some preclinical studies showed that the presence of KIF5B upstream of RET induces an increase of RET transcription, whereas the presence of other fusion partners, such as CCDC6, is associated with a lower degree of RET expression. This aspect could influence the response to RET inhibition,Citation38,Citation39 but more studies should be performed to address this issue. Moreover, KIF5B-RET fusion can strongly activate a signaling hub of various kinases, such as RET, EGFR, SRC, FGFR. This effect was not observed for other RET rearrangements (ie, CCDC6-RET and NCOA4-RET) and can be overcome through inhibiting both RET and EGFR, as Das and CaganCitation40 demonstrated in an in vitro study. These findings are corroborated in vivo, as reviewed by Ferrara et al.Citation23 Patients with KIF5B-RET fusion mainly achieved lower overall response rates than patients with other RET fusions.
Recently, through other in vitro experiments, some missense RET mutations have been found associated with resistance to cabozantinib, lenvatinib, and vandetanib in patients with medullary thyroid cancer. In particular, the authors found a number of TKI-resistant mutations located in the Gly-rich loop (L730, E732, and V738), the gatekeeper residue (V804), or the hinge strand (Y806, A807, and G810) that comprise about two-thirds of the drug binding pocket. They also demonstrated that different aminoacid substitutions of the same site could have different consequences for drug resistance, suggesting that not only the site of the kinase but also the type of amino acid should be considered when evaluating drug sensitivity.Citation41
However, to date these mutations have not been observed in tumor samples from patients with RET-rearranged or RET-mutant cancer that have developed an acquired resistance to RET inhibitors. Recently, a case report showed a RET-rearranged lung cancer patient that developed a secondary RET mutation (S904F) at the onset of resistance to vandetanib. The mutation was a serine-to-phenylalanine substitution at codon 904 in the activation loop of the RET kinase domain and conferred resistance to vandetanib by increasing the ATP affinity and autophosphorylation activity of RET kinase. This result indicates that missense mutations in the activation loop of the kinase domain are able to increase kinase activity and confer drug resistance through allosteric effects.Citation42 Another case report described the effect of V804M gatekeeper mutation on resistance to vandetanib.Citation43
As we know, in ALK rearranged patients, the frequency of ALK mutations as a resistance mechanism is higher in those patients receiving second generation ALK-inhibitors (alectinib or ceritinib), more potent than first generation inhibitors such as crizotinib,Citation44 suggesting that the potency of the drug in inducing a block of the target is associated with induction of resistance mutations on the target. This could suggest that more potent anti-RET agents could induce RET mutation, also indicating a more effective anti-RET activity, with respect to the current used agents.
Concurrent genomic alterations could also have a role in conferring resistance. Preclinical studies in a RET-rearranged lung cancer cell line showed that the EGFR-mediated signaling could mediate resistance to multikinase inhibition providing a rationale to cotarget EGFR to reduce the risks of developing drug resistance.Citation45,Citation46 In the study by Chang et al,Citation45 CCDC6-RET positive lung cancer cells were highly sensitive to RET inhibition, but EGFR signaling was responsible for resistance to sunitinib, vandetanib, and sorafenib, through the induction of ERK and AKT activity. Moreover, they demonstrated that endothelial cells, which are known to produce EGF, decreased the sensitivity of RET inhibitors. In addition, the results of Vaishnavi et al,Citation46 performed on different models of lung cancer cell lines carrying ALK, ROS1, RET, and NTRK1 fusions, confirm that EGFR signaling was involved at different levels in determining resistance to multikinase inhibitors, and that treatment with the EGFR-TKI gefinitb abrogated all the EGFR contributions.
Another potential resistance mechanism, both primary and acquired, is the MDM2 amplification. Pre- and post tumor biopsy were obtained from RET-rearranged lung cancer patients treated with cabozantinib, and amplification of the gene was observed in 50% of patients undergoing resistance, highlighting the possibility to combine anti-MDM2 agents with RET inhibitors.Citation47
Another potential resistant mechanism to RET inhibition, observed in in vitro studies, is the activation of the MAPK pathway. By studying RET-rearranged cell lines treated with ponatinib, cell clones resistant to the drug were obtained and the molecular characteristics of those was studied. Although cells retained the expression of the RET fusion, phosphorylation lacked. The activation of the MAPK signaling was seen in both cell lines, in one case due to the induction of NRAS mutation and in the other model due to the overexpression of EGFR and AXL.Citation48 These results should be confirmed in a clinical setting and, if confirmed, could open the possibility for combinational treatment using MAPK-inhibiting drugs ().
Table 1 Summary of known mechanisms of resistance to RET inhibition
Future prospects of specific RET inhibition
The results in terms of activity and tolerability of the various multikinase inhibitors, active but not specific against RET, led us to investigate a new anti-RET specific kinase inhibitor, RXDX-105. Its IC50 is 0.3, 0.3–0.8, and 5–15 nM against wild-type RET, RET rearrangements, and mutated RET, respectively. VEGFRs are spared from inhibition by this drug. It was evaluated in a phase I trial including 28 RET-fusion positive NSCLC patients. The most common G3 adverse events reached no more than 10% and no G4 toxicity was reported. None of the patients with KIF5B-RET fusion experienced a response, whereas among patients with non-KIF5B-RET fusions ORR was 75%, suggesting a relevant role of patient selection according to rearrangement type.Citation49
BLU-667 is a novel small-molecule RET inhibitor. It has been designed for high potency and selectivity against oncogenic RET alterations, including the most frequent RET rearrangements (eg, KIF5B–RET and CCDC6–RET). It was tested preclinically in RET-driven thyroid, lung, and colorectal cancers. KIF5B–RET autophosphorylation was inhibited by BLU-667 in vitro over 20-fold more potently than RXDX-105. Durable responses without significant off-target toxicity in patients with RET-altered NSCLC and medullary thyroid cancer prompted clinical investigation.Citation29 A phase one open-label first-in-human study was designed for BLU-667 in patients with medullary thyroid cancer, RET-altered NSCLC, and other RET-altered solid tumors, and has been recruiting currently (NCT03037385).Citation50
LOXO-292 is another highly selective ATP-competitive RET inhibitor, which has nanomolar potency against diverse RET alterations, including anticipated acquired resistance mutations. It has advantageous pharmacokinetic features, such as high bioavailability, significant penetration of central nervous system, and low potential for drug interactions.Citation30 Patients with advanced RET-altered tumors, treated with any prior multikinase inhibitors, including lung cancer, are under recruitment in a phase 1/2, open-label, first-in-human study with LOXO-292 (NCT03157128).Citation51 In the meantime, a study of patients treated with LOXO-292 showed a rapid clearance of RET variants in cell-free DNA ().Citation52
Table 2 Summary of new RET-specific drugs
Brain metastases in RET-rearranged NSCLC
Few studies reported information on the frequency, responsiveness, and overall outcomes in RET-rearranged advanced NSCLC patients with central nervous system (CNS) metastases. A recent paper by Drilon et alCitation53 focused on this topic. They showed that the frequency of CNS involvement in these patients is 25% at diagnosis, but lifetime prevalence can reach almost a half. Furthermore, the cumulative incidence of CNS lesions in RET-positive NSCLC patients is higher than ROS1-positive and lower than ALK-positive patients. They found a low intracranial response rate when these patients were treated with various multikinase inhibitors (alectinib, cabozantinib, ponatinib, sunitinib, vandetanib, vandetanib + everolimus): two of eleven patients (18%), one treated with alectinib and one with vandetanib + everolimus. In both these patients with responding CNS metastases, CCDC6-RET fusion was present. PFS and OS were short in patients with brain metastases: 2.1 months (95% CI =1.3–2.9 months) and 3.9 months (95% CI =1.9–5.4), respectively. However, these outcomes can echo the limited efficacy of multikinase inhibitors in RET-rearranged NSCLC patients. The combination of vandetanib and everolimus can represent an option to optimize blood–brain-barrier penetration as previously reported.Citation54 Some data about intracranial antitumor activity through the selective RET-directed inhibitors BLU-667 and LOXO-292 are emerging.Citation55,Citation56
RET fusions as a resistance mechanism to EGFR inhibition
Among NSCLC patients with activating EGFR mutations who undergo treatment with EGFR-TKIs, in around 15%– 20% the acquired resistance mechanisms remain unknown. The recent use of new methods of comprehensive genome profiling allowed us to identify some gene rearrangements as resistance mechanisms to EGFR-TKIs.
First, in 2015, some authorsCitation57 reported two cases of EGFR-mutant NSCLC patients treated with erlotinib, who developed CCDC6-RET fusion as detected via a hybrid-capture-based comprehensive genomic profiling assay in tumor tissue from rebiopsy. In another case, a retrospective analysis of the Foundation Medicine database allowed the identification of an acquired NCOA4-RET fusion in a NSCLC patient progressing on EGFR-TKI.Citation57
More recently, the Foundation Medicine database was explored to identify EGFR-mutant NSCLC patients. The tumor and blood samples were analyzed for BRAF or RTK fusions. In four patients, three RET fusions were found (ie, CCDC6-RET, NCOA4-RET, and TRIM24-RET). After the appearance of these RET rearrangements during an EGFR-TKI, RET inhibitors were delivered. One patient with CCDC6-RET fusion post-erlotinib had no benefit to single-agent alectinib. Another patient with NCOA4-RET fusion post-afatinib experienced stable disease through the combination of afatinib and cabozantib.Citation58
An in vitro model with EGFR-mutant lung cancer cell lines expressing CCDC6-RET showed that the combination of an EGFR-TKI with the selective inhibitor BLU-667 achieved a decrease in cell viability. In the same paper by Piotrowska et al,Citation59 some cases of EGFR-mutant NSCLC patients treated with combined EGFR and RET inhibition after the occurrence of RET fusions are reported. One patient with CCDC6-RET fusion post-afatinib was treated with erlotinib plus cabozantinib, but obtained no significant benefit. In two other patients, one with CCDC6-RET post-osimertinib and one with NCOA4-RET post-afatinib/cetuximab, the combination of osimertinib and BLU-667 achieved significant tumor response with marked tumor shrinkage.
These reports suggest that a selective RET inhibition combined with EGFR-TKI could help to manage acquired resistance to EGFR-TKIs when RET fusions are documented as a resistance mechanism. However, specific clinical trials are needed to recommend this as a standard approach.
Conclusion
The RET gene is one of the already known oncogenes undergoing activating rearrangements in a small subpopulation of lung cancer patients. The availability of multikinase inhibitors, active against RET among various targets, encouraged us to find a target therapy also for these patients. By using multikinase inhibitors to target RET, the consequent concomitant VEGFR and EGFR inhibitions lead to off-target toxicity. As a consequence these mutikinase inhibitors cannot be delivered at the dose necessary for RET inhibition. Both retrospective and prospective studies showed a good activity of some multikinase inhibitors in RET fusion positive NSCLC patients, but not sufficient to consider these drugs as a valid alternative to chemotherapy as achieved by EGFR- and ALK-inhibitors in other oncogene-addicted tumors. From these studies an intrinsic resistance emerged according to the type of RET fusion partners. Moreover, some acquired resistance mutations in rearranged RET were found during the treatment with multikinase inhibitors. Nowadays encouraging prospects derive from the development of selective RET inhibitors with high potency, but without off-target toxicity. Some early phase clinical trials are ongoing, giving the hope that soon new drugs will be available to specifically treat those 1%–2% of NSCLC patients with a RET rearrangement, sparing them from first-line chemotherapy.
Disclosure
The authors report no conflicts of interest in this work.
References
- NovelloSBarlesiFCalifanoRMetastatic non-small-cell lung cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-upAnn Oncol201627suppl 5v1v2727664245
- BronteGRizzoSLa PagliaLDriver mutations and differential sensitivity to targeted therapies: a new approach to the treatment of lung adenocarcinomaCancer Treat Rev201036Suppl 3S21S2921129606
- RolfoCMackPCScagliottiGVLiquid biopsy for advanced non-small cell lung cancer (NSCLC): a statement paper from the IASLCJ Thorac Oncol20181391248126829885479
- Cancer Genome Atlas Research NetworkComprehensive molecular profiling of lung adenocarcinomaNature2014511751154355025079552
- TakahashiMRitzJCooperGMActivation of a novel human transforming gene, RET, by DNA rearrangementCell19854225815882992805
- TakahashiMRet protooncogene and human-diseases – reviewInt J Oncol199441818421566893
- de GraaffESrinivasSKilkennyCDifferential activities of the RET tyrosine kinase receptor isoforms during mammalian embryogenesisGenes Dev200115182433244411562352
- ChiXMichosOShakyaRRet-dependent cell rearrangements in the Wolffian duct epithelium initiate ureteric bud morphogenesisDev Cell200917219920919686681
- AiraksinenMSSaarmaMThe GDNF family: signalling, biological functions and therapeutic valueNat Rev Neurosci20023538339411988777
- ArighiEBorrelloMGSariolaHRET tyrosine kinase signaling in development and cancerCytokine Growth Factor Rev2005164–544146715982921
- MarxSJAgarwalSKKesterMBMultiple endocrine neoplasia type 1: clinical and genetic features of the hereditary endocrine neoplasiasRecent Prog Horm Res19995439743810548885
- HendyGNKajiHCanaffLCellular functions of meninAdv Exp Med Biol2009668375020175451
- MarxSJMolecular genetics of multiple endocrine neoplasia types 1 and 2Nat Rev Cancer20055536737515864278
- EngCClaytonDSchuffeneckerIThe relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET Mutation Consortium AnalysisJAMA19962761915758918855
- CerratoAde FalcoVSantoroMMolecular genetics of medullary thyroid carcinoma: the quest for novel therapeutic targetsJ Mol Endocrinol200943414315519383830
- PrescottJDZeigerMAThe RET oncogene in papillary thyroid carcinomaCancer2015121132137214625731779
- SantoroMMelilloRMFuscoARET/PTC activation in papillary thyroid carcinoma: European Journal of endocrinology Prize LectureEur J Endocrinol2006155564565317062879
- JuYSLeeWCShinJYA transforming KIF5B and RET gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencingGenome Res201222343644522194472
- DacicSLuvisonAEvdokimovaVRET rearrangements in lung adenocarcinoma and radiationJ Thorac Oncol20149111812024346100
- WangRHuHPanYRET fusions define a unique molecular and clinicopathologic subtype of non-small-cell lung cancerJ Clin Oncol201230354352435923150706
- KatoSSubbiahVMarchlikEElkinSKCarterJLKurzrockRRET aberrations in diverse cancers: next-generation sequencing of 4,871 patientsClin Cancer Res20172381988199727683183
- ChaoBHBriesewitzRVillalona-CaleroMARET fusion genes in non-small-cell lung cancerJ Clin Oncol201230354439444123150705
- FerraraRAugerNAuclinEBesseBClinical and translational implications of RET rearrangements in non-small cell lung cancerJ Thorac Oncol2018131274529128428
- PlenkerDRiedelMBrägelmannJDrugging the catalytically inactive state of RET kinase in RET-rearranged tumorsSci Transl Med20179394eaah614428615362
- Plaza-MenachoIMologniLMcDonaldNQMechanisms of RET signaling in cancer: current and future implications for targeted therapyCell Signal20142681743175224705026
- MatsubaraDKanaiYIshikawaSIdentification of CCDC6-RET fusion in the human lung adenocarcinoma cell line, LC-2/adJ Thorac Oncol20127121872187623154560
- SaitoMIshigameTTsutaKKumamotoKImaiTKohnoTA mouse model of KIF5B-RET fusion-dependent lung tumorigenesisCarcinogenesis201435112452245625064355
- LiGGSomwarRJosephJAntitumor activity of RXDX-105 in multiple cancer types with RET rearrangements or mutationsClin Cancer Res201723122981299028011461
- SubbiahVGainorJFRahalRPrecision targeted therapy with BLU-667 for RET-driven cancersCancer Discov20188783684929657135
- SubbiahVVelchetiVTuchBBSelective RET kinase inhibition for patients with RET-altered cancersAnn Oncol20182981869187629912274
- GautschiOMiliaJFilleronTTargeting RET in patients with RET -rearranged lung cancers: results from the Global, Multicenter RET RegistryJ Clin Oncol201735131403141028447912
- DrilonARekhtmanNArcilaMCabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trialLancet Oncol201617121653166027825636
- LeeSHLeeJKAhnMJVandetanib in pretreated patients with advanced non-small cell lung cancer-harboring RET rearrangement: a phase II clinical trialAnn Oncol201728229229727803005
- YohKSetoTSatouchiMVandetanib in patients with previously treated RET-rearranged advanced non-small-cell lung cancer (LURET): an open-label, multicentre phase 2 trialLancet Respir Med201751425027825616
- HoriikeATakeuchiKUenamiTSorafenib treatment for patients with Ret fusion-positive non-small cell lung cancerLung Cancer201693434626898613
- LipsonDCapellettiMYelenskyRIdentification of new ALK and RET gene fusions from colorectal and lung cancer biopsiesNat Med201218338238422327622
- VelchetiVHidaTReckampKLPhase 2 study of lenvatinib (Ln) in patients (PTS) with Ret fusion-positive adenocarcinoma of the lungAnn Oncol201627suppl_61204PD
- KohnoTIchikawaHTotokiYKIF5B-RET fusions in lung adenocarcinomaNat Med201218337537722327624
- RichardsonDSGujralTSPengSAsaSLMulliganLMTranscript level modulates the inherent oncogenicity of RET/PTC oncoproteinsCancer Res200969114861486919487296
- DasTKCaganRLKIF5B-RET oncoprotein signals through a multi-kinase signaling hubCell Reports201720102368238328877471
- LiuXShenTMooersBHMHilbergFWuJDrug resistance profiles of mutations in the RET kinase domainBr J Pharmacol2018175173504351529908090
- NakaokuTKohnoTArakiMA secondary RET mutation in the activation loop conferring resistance to vandetanibNat Commun20189162529434222
- Dagogo-JackIStevensSELinJJEmergence of a RET V804M gatekeeper mutation during treatment with vandetanib in RET-Rearranged NSCLCJ Thorac Oncol20181311e226e22730368414
- GainorJFDardaeiLYodaSMolecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancerCancer Discov20166101118113327432227
- ChangHSungJHMoonSUKimHSKimJWLeeJSEGF induced RET inhibitor resistance in CCDC6-RET lung cancer cellsYonsei Med J201758191827873490
- VaishnaviASchubertLRixUEGFR mediates responses to small-molecule drugs targeting oncogenic fusion kinasesCancer Res201777133551356328428274
- SomwarRSmithRHayashiTMDM2 amplification (Amp) to mediate cabozantinib resistance in patients (Pts) with advanced RET -rearranged lung cancersJ Clin Oncol20163415_suppl9068
- Nelson-TaylorSKLeATYooMResistance to RET-Inhibition in RET-Rearranged NSCLC is mediated by reactivation of Ras/MAPK signalingMol Cancer Ther20171681623163328500237
- DrilonAELiuSDoebeleRLBA19A phase 1B study of RXDX-105, a VEGFR-sparing potent RET inhibitor, in RETi-naïve patients with Ret fusion-positive NSCLCAnn Oncol201728suppl_5
- ClinicalTrials.gov Identifier: NCT03037385 Available from: https://clini-caltrials.gov/ct2/show/NCT03037385Accessed September 26, 2018
- ClinicalTrials.gov Identifier: NCT03157128 Available from: https://clini-caltrials.gov/ct2/show/NCT03157128Accessed September 26, 2018
- BesseBSubbiahVDrilonA105 Detection and clearance of RET variants in plasma cell free DNA (cfDNA) from patients (PTS) treated with LOXO-292Ann Oncol201829suppl_8ESMO2018
- DrilonALinJJFilleronTFrequency of brain metastases and multikinase inhibitor outcomes in patients with RET-rearranged lung cancersJ Thorac Oncol201813101595160130017832
- SubbiahVBerryJRoxasMSystemic and CNS activity of the RET inhibitor vandetanib combined with the mTOR inhibitor everolimus in KIF5B-RET re-arranged non-small cell lung cancer with brain metastasesLung Cancer201589767925576294
- DrilonAESubbiahVOxnardGRA phase 1 study of LOXO-292, a potent and highly selective RET inhibitor, in patients with RET -altered cancersJCO20183615_suppl102
- SubbiahVTaylorMLinJHighly potent and selective RET inhibitor, BLU-667, achieves proof of concept in a phase I study of advanced, RET-altered solid tumorsAACR Meet Abstr Online2018Abstract CT043
- KlempnerSJBazhenovaLABraitehFSEmergence of RET rearrangement co-existing with activated EGFR mutation in EGFR-mutated NSCLC patients who had progressed on first- or second-generation EGFR TKILung Cancer201589335735926187428
- SchrockABZhuVWHsiehWSReceptor tyrosine kinase fusions and BRAF kinase fusions are rare but actionable resistance mechanisms to EGFR tyrosine kinase inhibitorsJ Thorac Oncol20181391312132329883838
- PiotrowskaZIsozakiHLennerzJKLandscape of acquired resistance to osimertinib in EGFR-mutant NSCLC and clinical validation of combined EGFR and RET inhibition with osimertinib and BLU-667 for acquired RET fusionCancer Discov20188121529153930257958