
Abstract
We report a case of May–Hegglin anomaly (MHA) in a woman who had a successful labor and delivery under epidural anesthesia. MHA is an inherited thrombocytopenia easily misdiagnosed as idiopathic (immune) thrombocytopenic purpura (ITP). Early and appropriate diagnosis of MHA during pregnancy is essential for optimal maternal and neonatal delivery outcome. Additionally, it can avoid unnecessary diagnostic studies, such as bone marrow aspiration and biopsy, and even harmful therapies with corticosteroids, immunosuppressive agents, and splenectomy. Consequently, the most serious impacts of this disease are iatrogenic managements due to misdiagnosis. It seems that in patients with MHA, adequate clinical coagulation is far more dependent on adequate platelet function than any particular platelet count. The diagnosis of MHA may pose a challenge for clinicians managing pregnant women with thrombocytopenia.
Introduction
May–Hegglin anomaly (MHA) is a rare hematological disorder, inherited as an autosomal dominant trait, within the family of myosin heavy chain (MHC) disorders, including Fechtner syndrome, Sebastian syndrome, Epstein syndrome, and Alport-like syndromes.Citation1–Citation4 This group is also known as MYH9-related disorders (MYH9RDs), because all have largely overlapping phenotypes and result from mutations in the MYH9 gene on chromosome 22, which encodes the nonmuscle myosin heavy chain-IIA (NMMHC-IIA) protein.Citation3–Citation5 NMMHC-IIA is a cytoplasmic protein with expression in many tissues, including platelets.Citation1–Citation4
MYH9RDs are characterized by varying degrees of thrombocytopenia, giant platelets, and large (2–5 μm), well-defined, basophilic, cytoplasmic inclusion bodies (resembling Döhle bodies) in the granulocytes.Citation1–Citation5 These inclusion bodies result from deposition of MHC within white blood cells. Interestingly, they are not seen in platelets, and their presence in leukocytes can differentiate MHA from idiopathic (immune) thrombocytopenic purpura (ITP).Citation1–Citation5
Top of Form
MYH9RDs are considered very rare. The Italian Registry for MYH9RD indicates that the prevalence of the disorder in Italy is at least 3:1,000,000.Citation6 Because mild forms are discovered incidentally and severe forms are often misdiagnosed or underreported as other disorders, the actual prevalence is expected to be higher. MYH9RD has been diagnosed worldwide, and there is no evidence of variation in prevalence across ethnic populations.Citation7 In general, the worldwide incidence of MHA is unknown.
Thrombocytopenia occurs in approximately 50% of the patients with MHA, and the clinical manifestations vary from mild bleeding not requiring specific treatment up to severe bleeding episodes following trauma or surgery that require blood products.Citation1–Citation3,Citation8 However, despite severe thrombocytopenia in most patients, platelet function is frequently normal. Therefore, they are asymptomatic, discovered incidentally.Citation1–Citation3,Citation8
Thrombocytopenia is common during pregnancy; however, MHA is very rare, with 40 cases reported in the literature.Citation9 Diagnosis has been established prior to pregnancy in some women, but in the majority, the problem is first identified incidentally during pregnancy as thrombocytopenia on routine booking blood tests. Most of such cases, without careful inspection of blood smears and a thorough family and bleeding history, are initially misdiagnosed as refractory-to-treatment ITP.Citation9 Consequently, the most serious impacts of this disease are iatrogenic managements due to misdiagnosis.Citation3 The diagnosis of MHA may pose a challenge for clinicians managing pregnant women with thrombocytopenia. Herein, we report a case of MHA in a woman who had a successful labor and delivery under epidural anesthesia. The patient provided written informed consent for her data to be included in this case report.
Case description
The history of the patient starts 10 years ago on April 2006, when she presented as primigravida at 36 weeks gestation, booked for antenatal investigations. Patient was not known to have any medical illness or history of previous surgical procedures. Her platelet count was found to be 22×109/L, and she was admitted as a case of possible ITP. Coagulation profile was within normal limits, and there was no history of bleeding tendency. Family history positive for bleeding disorders was not reported. She was started on steroid therapy (initially with prednisolone and later with dexamethasone); however, the platelet count decreased to 19×109/L. After transfusion of 6 units of platelets, her platelet count was 20×109/L. Then, she received four cycles of rituximab (monoclonal antibody) and three doses of immunoglobulin, without response. Bone marrow biopsy revealed thalassemia trait and iron deficiency. Megakaryocytes and erythroblasts exhibited significant dysplasia. Differential diagnosis was between ITP and myelodysplastic syndrome. She had an uncomplicated cesarean section under general anesthesia, due to failure to labor progress. She received 18 units of platelets and 6 units of cryoprecipitate since the cesarean section. No bleeding tendency was observed perioperatively. Postoperatively, her platelet count was 65×109/L, with normal coagulation profile. She was discharged without any complication.
After 3 months, a second bone marrow biopsy did not reveal additional pathology and confirmed the initial diagnosis (ITP). Her platelet count was 18×109/L, so the patient was scheduled for splenectomy due to refractory ITP. In August 2006, she was transfused with 8 units of platelets and underwent an uncomplicated laparoscopic splenectomy under general anesthesia, with minimal blood loss. The spleen pathology report was consistent with the clinical diagnosis of ITP. After splenectomy, she had a platelet count 8×109/L. Two months later, she showed improvement to 50×109/L and, at 6 months, to 100×109/L. However, then her platelet count decreased again and ranged between 7 and 20×109/L.
In December 2007, the patient was then evaluated while she was on her second pregnancy at 2 months’ gestation. Her platelet count was 7×109/L, and she was advised admission and the need for treatment. However, she signed against medical advice. She was transfused with 6 units of platelets and received intravenous (IV) immunoglobulin 55 g (1 g/kg) and then was discharged with platelet count 45×109/L and on oral treatment with prednisolone. In June 2008, she had an uncomplicated labor and delivery that did not need any anesthetic management. During regular follow-up evaluations, the patient presented with hypothyroidism and started treatment with oral thyroxine.
About 2 years later, in January 2011, during regular follow-up evaluations, microscopic assessment of a peripheral blood smear showed giant platelets and cytoplasmic inclusion bodies in the granulocytes. Therefore, the patient was diagnosed as suffering from MHA. Hemoglobin electrophoresis revealed findings consistent with alpha thalassemia trait and sickle cell trait with iron deficiency anemia. The patient had another bone marrow examination, which revealed that she did not have ITP and that her thrombocytopenia was due to MYH9 gene-related hereditary macrothrombocytopenia. During her further course, she had two more pregnancies in 2011 and 2012, with spontaneous abortions. Her platelet count ranged between 8 and 20×109/L.
In 2014, the patient presented for labor and delivery after her fifth pregnancy, with spontaneous rupture of membrane at 39 weeks’ gestation. Her platelet count was 13×109/L, and she was transfused with 6 units of platelets, which increased the platelet count to 24×109/L. The hematology consult was that the patient should be treated as a patient with a normal coagulation status and did not require any other specific treatment. Additionally, a neuraxial blockade could be performed if needed. Once requested, an epidural catheter was easily inserted at L1–L2. The patient had an uncomplicated delivery and an uneventful hospital stay. She was discharged after 6 days.
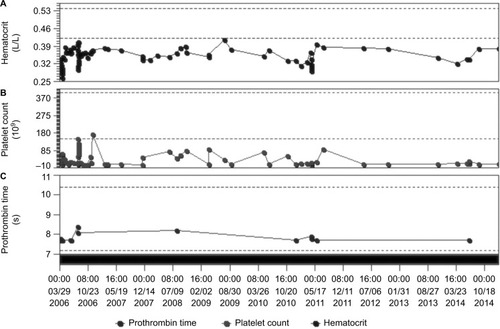
shows a graphic over a period of 9 years (2006–2014), presenting the range of changes in platelet count, prothrombin time, and hematocrit of the patient. Platelet count was transiently increased only after massive platelet transfusions, while most of the time, it ranged between 10 and 20×109/L. At the same time, no significant changes were noted in prothrombin time and hematocrit, except during labor and deliveries.
Figure 1 Graphic representation of a period of 9 years (2006–2014), presenting the range of changes in (A) hematocrit, (B) platelet count, and (C) prothrombin time.
Discussion
We have presented a case of MHA in a woman who underwent a successful labor and delivery under epidural anesthesia. Review of literature revealed that neuraxial blockade has been rarely reported in the past in MHA cases. Spinal anesthesia for cesarean section has been applied in four cases and epidural analgesia applied for labor and delivery in five cases.Citation10–Citation13
Interestingly, there is still no consensus regarding the absolute lower limit of platelet count that confers safety for neuraxial anesthesia. Even the very recent (2016) guidelines for obstetrical anesthesia from the American Society of Anesthesiologists (ASA)Citation14 avoid suggesting a clear guidance and just state: “A specific platelet count predictive of neuraxial anesthetic complications has not been determined.” In general, a platelet count of ≥50×109/L is recommended for safe delivery.Citation4 Therefore, in most cases, general anesthesia is preferred, due to severe thrombocytopenia and considering the risk of possible spinal or epidural hematoma.Citation14–Citation16
MYH9RDs result from mutations in the MYH9 gene. Depending on the position of the causative mutation within the gene, the risk increases for syndromic clinical manifestations.Citation3,Citation17 Bleeding diathesis, high-tone hearing loss, glomerular nephropathy, and presenile cataract are the clinical features of MYH9RDs in descending order of frequency.Citation5 Mutations in the neck region of the NMMHC-IIA protein are more likely associated with these comorbidities than mutations in the N- or C-terminal part of the gene.Citation3,Citation17 To date, at least 49 mutations of the MYH9 gene have been identified.Citation6 More specifically, MHA is caused by abnormalities in the MYH9 gene located on chromosome 22q12–13 and encoding NMMHC–IIA.Citation1–Citation4 The diagnosis of MHA has been conventionally based on morphological criteria alone, by microscopic assessment of a peripheral blood smear after conventional staining (such as May-Grünwald-Giemsa staining), demonstrating a triad of giant platelets, thrombocytopenia, and inclusions in the cytoplasm of leukocytes.Citation1–Citation4 It is important to note that electronic cell counters work mainly based on the size and therefore they often classify very large platelets as erythrocytes. As a consequence, in MYH9RD, these instruments greatly underestimate mean platelet volume, as well as platelet count.Citation18,Citation19
The pathogenesis of MHA is poorly understood. Reported platelet counts in MHA have ranged from less than 10×109/L to normal levels, indicating variable expressivity; however, the platelet structure, function, and life span are usually normal.Citation20 Additionally, no correlation was found between bleeding tendency and platelet count.Citation2 A qualitative defect of platelets may be responsible for mild bleeding diathesis even in the absence of thrombocytopenia, while severe bleeding results from both qualitative and quantitative platelet defects. Bone marrow examination shows normal megakaryocytes. The cause of the thrombocytopenia in MHA is thought to be defective megakaryocyte maturation and fragmentation, which may account for production of large platelets.Citation21
Thrombocytopenia is common during pregnancy. The most common causes include gestational thrombocytopenia, preeclamptic disorders of pregnancy and ITP. However, MHA is rare during pregnancy.Citation9 Whenever a family history of thrombocytopenia is absent or unclear, evaluation of peripheral blood slides is a simple and effective tool to distinguish patients with MHA from those with ITP, as platelets are significantly larger in patients with MHA than in those with ITP. In particular, a mean platelet diameter >3.74 μm distinguishes MHA from ITP with 86% sensitivity and 87% specificity.Citation22
The clinical spectrum of congenital thrombocytopenias in women ranges from severe bleeding tendency, recognized within the first few weeks of life, to mild conditions that may remain undetected even in adulthood. Positive bleeding history may include heavy menstrual bleeding, easy bruising, nose bleedings, bleeding after tooth extraction, and gum bleeding. Clinically, most women with MHA are asymptomatic, discovered incidentally, and require no specific treatment. In a recent systematic review of MHA during pregnancy, it was reported that 33% of women had no history of any bleeding symptoms.Citation9
As the MHA is an autosomal dominant characteristic, the fetus has 50% chance of inheriting the pathogenic variant and presenting thrombocytopenia and MHA.Citation1–Citation5,Citation23 Therefore, vaginal deliveries in women with severe thrombocytopenia should be considered at increased risk for neonatal intracranial bleeding.Citation1,Citation4 Approximately 65% of individuals diagnosed with MYH9RD have an affected parent.Citation3–Citation5 Prenatal diagnosis for pregnancies at increased risk is possible if the pathogenic variant in the family is known.Citation4 The definitive diagnosis of MHA is limited to those with inclusion bodies in granulocytes, due to aggregates of abnormal NMMHC-IIA, and no other organ dysfunction.Citation8 Genetic analysis can be used if the evidences are nondiagnostic, but access to this test is generally only available through specialist molecular testing facilities.
Differentiating between inherited and acquired thrombocytopenias may be difficult, and significant numbers of patients with MHA have been misdiagnosed with refractory ITP.Citation3,Citation24,Citation25 In most cases, the first presentation is identified during pregnancy as thrombocytopenia during routine antenatal blood tests, with incidental finding of low platelet count. The biggest risk for these patients is that they are subjected to unnecessary, ineffective, and potentially harmful treatments based on the misdiagnosis of ITP.Citation3 Consequently, the most serious impacts of this disease are iatrogenic managements due to misdiagnosis, and many of such patients suffer a lot before the final MHA diagnosis is established. Like in our case, a common scenario includes ineffective immunosuppressive (steroids) and intravenous immunoglobulin-G treatment, unnecessary repeated platelet and cryoprecipitate transfusions, multiple painful bone marrow examinations, and eventually ineffective, unnecessary, and potentially harmful splenectomy for steroid-resistant ITP.Citation26,Citation27
Routine transfusions of platelet concentrates are used in patients with MHA, to transiently increase platelet count. However, this is unnecessary and carries associated risks of possible alloimmunization, producing antibodies against human leukocyte antigen (HLA) and subsequent refractoriness to platelet infusions.Citation3–Citation5 Therefore, platelets and blood should be ready available for use; however, they should be administered only if abnormal active bleeding occurs, which cannot be otherwise managed.Citation4,Citation28 When available, transfusing platelets from HLA-matched donors should be preferred, as this reduces the risk of alloimmunization.Citation4,Citation28
There is no known prevention or treatment for the non-hematopoietic consequences of MHA. Previous therapy with steroids and/or high-dose immunoglobulins had no effect on platelet count or bleeding diathesis.Citation3–Citation5,Citation9 Hemostatic prophylaxis for labor and delivery may include desmopressin (DDAVP®), tranexamic acid, and cryoprecipitate transfusion.Citation4,Citation29 Preoperative use of desmopressin can be considered, because it has been proven to reduce the bleeding tendency in many acquired and congenital disorders of platelet function, including MHA.Citation4,Citation8,Citation29 An assessment of the bleeding risk should be performed during the third trimester of pregnancy. Appropriate hemostatic cover can then be arranged to minimize the risk of primary postpartum hemorrhage. Women with no bleeding history and platelet count of at least 80–109×109/L are at low risk for bleeding and do not require platelet transfusion for labor and delivery.Citation4 Desmopressin and/or tranexamic acid are generally adequate as hemostatic cover.Citation4,Citation29 However, women with a positive bleeding history or with platelet count less than 50×109/L may additionally require prophylactic platelet transfusion. Eventually, splenectomy, which is a treatment for refractory ITP, has not shown any improvement in the platelet count or any decrease in the bleeding tendency.Citation26,Citation27 Consequently, splenectomy is contraindicated in all hereditary macrothrombocytopenias, including MHA.Citation26,Citation27
Conclusion
MHA is an inherited thrombocytopenia that is easily misdiagnosed as ITP, if careful inspection of blood smear and family history are overlooked. Early and appropriate diagnosis of MHA during pregnancy is essential for optimal maternal and neonatal delivery outcome. Additionally, this can avoid unnecessary diagnostic studies, such as bone marrow aspiration and biopsy, and even harmful therapies with corticosteroids, immunosuppressive agents, and splenectomy. It seems that in patients with MHA, adequate clinical coagulation is far more dependent on adequate platelet function than any particular platelet count. A qualitative defect of platelets may be responsible for mild bleeding diathesis even in the absence of thrombocytopenia, while severe bleeding results from both qualitative and quantitative platelet defects. MHA should be suspected whenever a patient has a low platelet count or a bleeding diathesis of unknown origin. Early and appropriate diagnosis of MHA during pregnancy is essential for optimal maternal and neonatal delivery outcome.
Disclosure
The authors report no conflict of interest in this work.
References
- SaitoHKunishimaSMay-Hegglin anomalyAm J Hematol20088330430617975807
- NorisPSpediniPBellettiSMagriniUBalduiniCLThrombocytopenia, giant platelets, and leukocyte inclusion bodies (May-Hegglin anomaly): clinical and laboratory findingsAm J Med199810443553609576409
- AlthausKGreinacherAMYH9-related platelet disordersSemin Thromb Hemost200935218920319408192
- SavoiaAPecciAMYH9-related disordersGeneReviews® [Internet] [updated July 16, 2015]. Available from https://www.ncbi.nlm.nih.gov/books/NBK2689/Accessed January 12, 2017
- BalduiniCLPecciASavoiaARecent advances in the understanding and management of MYH9-related inherited thrombocytopeniasBr J Haematol2011154216117421542825
- SaposnikBBinardSFenneteauOFrench MYH9 networkaMutation spectrum and genotype-phenotype correlations in a large French cohort of MYH9-related disordersMol Genet Genomic Med20142429731225077172
- Registro Italiano della Malattia MYH9-correlata [homepage on the Internet] [updated June 2014]. Available from: http://www.registromyh9.org/Accessed January 12, 2017
- SehbaiASAbrahamJBrownVKPerioperative management of a patient with May-Hegglin anomaly requiring craniotomyAm J Hematol200579430330816044442
- HusseinBAGomezKKadirRAMay-Hegglin anomaly and pregnancy: a systematic reviewBlood Coagul Fibrinolysis201324555456123811802
- FishmanEBConnorsJMCamannWRAnesthetic management of seven deliveries in three sisters with the May-Hegglin anomalyAnesth Analg200910851603160519372343
- DuffPJacksonMTPregnancy complicated by rhesus sensitization and the May-Hegglin anomalyObstet Gynecol1985653 suppl7S10S3919350
- KotelkoDMAnaesthesia for caesarean delivery in a patient with May-Hegglin anomalyCan J Anaesth1989363 pt 13283302720871
- NelsonLHDewanDMMandellGLObstetric and anesthetic considerations in the May-Hegglin anomaly. A case reportJ Reprod Med19933843113138501742
- Practice guidelines for obstetric anesthesia: an updated report by the American Society of Anesthesiologists Task Force on Obstetric Anesthesia and the Society for Obstetric Anesthesia and PerinatologyAnesthesiology201612427030026580836
- TakabayashiRNishikidoONaganoKAnesthetic management for cesarean delivery in a patient with May Hegglin anomalyMasui2007561198119917966627
- Garcia VallejoGCabellosMKabiriMFraileJRCuestaJAnaesthetic implications in a pregnant patient with an extreme thrombocytopenia due to a May-Hegglin anomaly: general or regional anaesthesia?Rev Esp Anestesiol Reanim20146146046524704095
- PecciAPanzaEPujol-MoixNPosition of nonmuscle myosin heavy chain IIA (NMMHC-IIA) mutations predicts the natural history of MYH9-related diseaseHum Mutat200829340941718059020
- AlthausKGreinacherAMYH9-related platelet disordersSemin Thromb Hemost200935218920319408192
- UratoACRepkeJTMay-Hegglin anomaly: a case of vaginal delivery when both mother and fetus are affectedAm J Obstet Gynecol199817912602619704798
- HamiltonRWShaikhBSOttieJNPlatelet function, ultrastructure and survival in the May-Hegglin anomalyAm J Clin Pathol1980746636687446470
- GoodwinHAGinsburgADMay–Hegglin anomaly: a defect in megakaryocyte fragmentation?Br J Haematol1974261171274853110
- NorisPBiinoGPecciAPlatelet diameters in inherited thrombocytopenias: analysis of 376 patients with all known disordersBlood20141246e4e1024990887
- SeriMPecciADi BariFMYH9-related disease: May-Hegglin anomaly, Sebastian syndrome, Fechtner syndrome, and Epstein syndrome are not distinct entities but represent a variable expression of a single illnessMedicine20038220321512792306
- BalduiniCLNorisPInherited thrombocytopeniasHematology20152037437526084367
- BalduiniCLPecciANorisPDiagnosis and management of inherited thrombocytopeniasSemin Thromb Hemost201339216117123397552
- DrachmanJGInherited thrombocytopenia: when a low platelet count does not mean ITPBlood2004103239039814504084
- BalduiniCLDrachmanJGRole of splenectomy in inherited thrombocytopeniasBlood2004104122715294857
- ChabaneHGallaisYPathierDTcherniaGGaussemPDelivery management in a woman with thrombocytopenia of the May–Hegglin anomaly typeEur J Obstet Gynecol Reprod Biol20019912412511604201
- RosenACHafnerESternsiteWKierPKyrleADas May-Hegglin-Syndrom in der Schwangerschaft. [Das May-Hegglin-Syndrom in der Schwangerschaft]Geburtshu Frauenheilk199757301312 German