
Abstract
Although the concept of equivalence for drugs (generics) and biologics (biosimilars) has been readily adopted, the concept of equivalence or indistinguishable characteristics for class III medical devices has yet to be specifically addressed regarding a defined regulatory approval process in the US. In September 2015, GenVisc 850® (sodium hyaluronate), a hyaluronic acid approved for the treatment of knee osteoarthritis, was approved by the US Food and Drug Administration (FDA) based upon indistinguishable characteristics in comparison to an approved branded hyaluronic acid (Supartz®/Supartz FX™). The purpose of this paper is to review the methodology and report the main outcomes used to demonstrate clinical comparability of GenVisc 850 with Supartz/Supartz FX. The FDA approval was collectively attained using prospectively defined methods for preclinical, physical, and chemical testing, as well as noninferiority in clinical performance comparisons. Evidence from five randomized controlled studies of Supartz/Supartz FX vs saline control injections (used for Supartz approval), two randomized controlled trials of GenVisc 850 vs saline control injections, and one randomized controlled study of GenVisc 850 vs Supartz/Supartz FX provided evidence of safety for GenVisc 850. Efficacy was further assessed based on assessment of the same Supartz studies and three prospectively identified GenVisc 850 studies. A Bayesian network meta-analysis was used to demonstrate that the clinical efficacy of GenVisc 850 was noninferior to Supartz/Supartz FX and superior to saline control. Overall, safety of GenVisc 850 was similar to that of Supartz/Supartz FX and saline control injections, while efficacy of GenVisc 850 was noninferior to that of Supartz/Supartz FX and superior to saline control injections.
Introduction
Since the enactment of the Drug Price Competition and Patent Term Restoration Act of 1984,Citation1 >10,000 generic drugs have entered US market. Generic drugs now account for 88% of prescriptions dispensed in the USCitation2 with generic substitution responsible for >$239 billion in annual health-care savings.Citation3 More recently, the Biologics Price Competition and Innovation Act of 2009,Citation4 enacted as part of health-care reform (Affordable Care Act), defined an abbreviated licensure pathway for biological products shown to be biosimilar to a Food and Drug Administration (FDA)-licensed reference product. A biological product is a virus, therapeutic serum, toxin, antitoxin, vaccine, blood, blood component or derivative, allergenic product, or analogous product intended to prevent, treat, or cure a medical condition. The biosimilar pathway allows a manufacturer to demonstrate equivalence to a branded or reference product based on establishing an identical mechanism of action, route of administration, composition, dosage form and strength, and indication for use. In March 2015 utilizing this guidance, the FDA approved filgrastimsndz (Sandoz International GmbH, Holzkirchen, Germany), an injectable granulocyte colony-stimulating factor, as the first generic biosimilar in US pharmaceutical history.Citation5 According to an analysis by the Congressional Budget Office, the continued availability of new biosimilars is projected to save $25 billion in health-care costs over the next 10 years.Citation6
For medical devices, the 510(k) route – reserved for class II devices (moderate-to-high risk) – was introduced in 1976 to establish a pathway for determining substantial equivalence of a device to a predicate device already on the market. Since inception, the FDA has cleared >140,000 devices via the 510(k) program, allowing a pathway to commercialization for substantially equivalent class II medical devices and allowing for significant competition in the market. To date, a similar pathway for class III (high risk) medical device approval has yet to be adopted by the FDA, although a viable regulatory pathway was established ~20 years ago through the FDA Modernization Act.Citation7
Intra-articular injections of hyaluronic acid (HA), commonly referred to as viscosupplementation, have been classified in the US by the FDA as class III medical devices for >20 years based on the presumption that the primary mechanism of action in alleviating pain is due to viscoelastic or mechanical properties of HA in solution rather than biological activity, with eleven products receiving FDA approval for the treatment of knee osteoarthritis (OA) since 1997, including Supartz®/Supartz FX™ (sodium hyaluronate; Bioventus LLC, Durham, NC, USA). In 1995, GenVisc 850® (sodium hyaluronate; OrthogenRx, Inc., Doylestown, PA, USA) was approved as a generic drug to Supartz/Artz/Artzal in Japan and is currently approved in >60 countries with extensive postmarketing safety data in >35 million doses administered to patients. The active components of GenVisc 850 (known under the branded name of Adant® outside the US) were specifically formulated and demonstrated to be equivalent in chemical composition, molecular characteristics, and dosage form to Supartz/Supartz FX. In September 2015, GenVisc 850 was approved by US FDA as a class III medical device based upon indistinguishable physical, chemical, and clinical (noninferiority) performance compared with the approved product, Supartz/Supartz FX, as well as documented superior performance compared with saline control injections. GenVisc 850 also demonstrated no unexpected safety risks, including demonstrated safety in the largest and longest duration saline-controlled clinical study of HA for knee OA.Citation8 The purpose of this paper is to review the prospectively defined methodology, analyses, and outcomes that were utilized to demonstrate similar clinical performance and attain US marketing approval for GenVisc 850.
Characteristics of GenVisc 850
GenVisc 850 is a viscoelastic nonpyrogenic solution of purified, high-molecular-weight sodium hyaluronate with a pH of 6.8–7.8. The sodium hyaluronate is a polysaccharide containing repeating disaccharide units of glucuronic acid and N-acetylglucosamine derived from a bacterial fermentation process. The average molecular weight is 850 kDa with a range of 620–1,170 kDa (hence the designation GenVisc 850). Each 2.5 mL solution contains 10 mg/mL sodium hyaluronate dissolved in 1% physiological saline. GenVisc 850 is indicated for the treatment of knee OA pain in patients who have failed to respond adequately to conservative nonpharmacological therapy and simple analgesics. The characteristics and indications for use of GenVisc 850 are indistinguishable from those of Supartz/Supartz FX.Citation9
Preclinical and analytical evaluation
Preclinical studies were conducted with GenVisc 850 to evaluate analytical chemistry; rheological and molecular weight dispersion; bioequivalence; acute, subacute, and chronic toxicity; mutagenicity/genotoxicity; immunogenicity/sensitization; irritation; hemolysis; release specifications; and sterilization validation. Overall, these tests confirmed that the chemical attributes, analytical characteristics, and rheological (molecular chain interactions) and molecular weight dispersion characteristics of multiple lots of GenVisc 850 were indistinguishable from various lots of the approved medical device, Supartz/Supartz FX. As part of this testing, 15 blinded samples of batches of GenVisc 850 or Supartz/Supartz FX were submitted to a testing laboratory, analyses were conducted under Good Laboratory Practices, and the samples were determined to be indistinguishable.Citation10 Furthermore, it was observed that the intraproduct variation was not different than the interproduct variation, using these same parameters.
Clinical standards for comparability evaluation
GenVisc 850 safety evaluation
Safety methods
Given that GenVisc 850 and Supartz/Supartz FX were formulated to have comparable attributes that were confirmed by analytical testing, the primary question arises as to whether such testing can translate to comparable clinical performance. The comprehensive evidence supporting safety of GenVisc 850 is summarized in the clinical results published in both the GenVisc 850 and the Supartz/Supartz FX Summary of Safety and Effectiveness Data,Citation9,Citation11 where clinical safety was evaluated in five prospective, randomized, double-blind studies of Supartz/Supartz FX (n=619) vs saline control (n=537) injections.Citation11 Additional evidence of a comparable safety profile for GenVisc 850 was provided by a multicenter, double-blind, randomized trial that compared GenVisc 850 (n=116) with Supartz/Supartz FX (n=113) in patients with knee OA.Citation12
Safety results
In the five studies of Supartz/Supartz FX vs saline control, the most common adverse events (AEs) were arthralgia, arthropathy, back pain, nonspecific pain, injection site reaction, headache, and injection site pain (). There were no statistically significant differences in the incidence of AEs between Supartz/Supartz FX and control groups. This integrated analysis demonstrated that, in relation to saline control, Supartz/Supartz FX had a comparable safety profile.
Table 1 Adverse events reported in five randomized controlled trials with Supartz/Supartz FX vs saline control injections
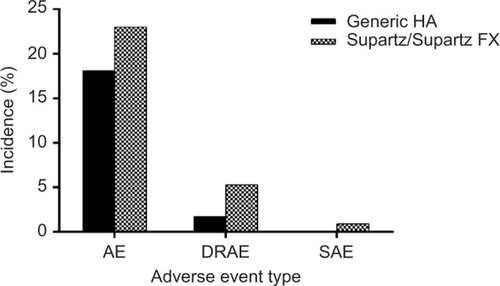
In the multicenter, double-blind, randomized trial comparing GenVisc 850 with Supartz/Supartz FX, the incidence of AEs was 18.1% vs 23.0% (P=0.36) for any AE, 1.7% vs 5.3% (P=0.17) for device-related AEs, and 0% vs 0.9% (P=1.0) for serious AEs with GenVisc 850 and Supartz/Supartz FX, respectively, after 6 weeks (). The serious AE reported in the Supartz/Supartz FX group was prostatic hyperplasia treated with surgical excision, which was deemed unrelated to the device.
Figure 1 Adverse event incidence in a randomized study of GenVisc 850 vs Supartz/Supartz FX.
Abbreviations: AE, adverse event; DRAE, device-related adverse event; HA, hyaluronic acid; SAE, serious adverse event.
Safety conclusion
The totality of evidence from five randomized controlled trials of Supartz/Supartz FX vs saline control injections and a randomized controlled trial of GenVisc 850 vs Supartz/Supartz FX provided adequate evidence of safety for FDA approval of GenVisc 850. Safety was further supported by the postmarketing safety reports of Supartz/Supartz FX over the past 20 years.
GenVisc 850 clinical efficacy evaluation
Efficacy methods
Clinical efficacy of the GenVisc 850 was evaluated using several separate comparative methodologies and approaches. The results of a randomized controlled trial and Bayesian meta-analysis of all available relevant studies were used to show that the clinical performance of GenVisc 850 is similar to Supartz and superior to PBS. A randomized controlled, multicenter clinical trial compared GenVisc 850 with Supartz, while Bayesian analyses evaluated the noninferiority of GenVisc 850 using data from randomized controlled trials that studied GenVisc 850 and Supartz. A first Bayesian analysis used only data from four studies that collected Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) A pain score.Citation16 A second analysis added Karlsson studyCitation18, which used WOMAC A as outcome, however, included patients with a Kellgren–Lawrence score of 4. Additional analyses were carried out on data from nine studies (five that collected WOMAC A pain score and four that did not) and the second analysis. In these analyses, if studies had WOMAC A it was used, if not then the Lequesne score (if available) was converted into WOMAC A by multiplying by a factor of 7 to the mean change from baseline and the standard deviation (SD).
A prospectively defined Bayesian network meta-analysis using random effects was utilized for combining direct and indirect evidence using noninformative prior distributions.Citation13,Citation14 Posterior inferences were carried out by sampling from the posterior distribution of the parameters using Gibbs sampling in OpenBUGS,Citation15 where 50,000 samples are obtained from the posterior distribution after the initial 5,000 are discarded. To decrease autocorrelation, only every tenth sample was used in calculations. Mean, SD, and 95% credible interval were obtained for each marginal posterior distribution. Convergence of the Markov chain Monte Carlo sampler was checked using the Brooks–Gelman–Rubin method. Convergence of the sampler was established if the ratio of within-chain and between-chain variability for multiple chains starting at different initial values was close to 1. With our specifications, this was established after the 5,000 burn-in samples.
We hypothesized that the efficacy of GenVisc 850 and Supartz/Supartz FX was each superior to saline control and that GenVisc 850 would be noninferior to Supartz/Supartz FX. Since Supartz/Supartz FX was already approved by the FDA, documenting the superiority over saline control using this methodology would serve to validate the approach. A priori, a 4 mm difference was established as the noninferiority margin comparing GenVisc 850 with Supartz/Supartz FX.
Efficacy results
In the five prospective, double-blind, randomized trials of Supartz/Supartz FX vs saline control, there was a statistically greater reduction in total Lequesne IndexCitation16 score of 2.7 for Supartz/Supartz FX vs 2.0 for saline control (P=0.003), thereby confirming the approval conclusions of the FDA in considering the Premarket Approval (PMA) application for Supartz/Supartz FX and supporting the statistical approach in this Bayesian analysis. Furthermore, in an analysis of a randomized controlled trial of GenVisc 850 vs Supartz/Supartz FX, mean reduction in pain on movement >6 weeks assessed with a 100 mm visual analog scale was 50±21 in each group, while mean improvement in total Lequesne Index score was 5.7±3.2 for GenVisc 850 and 6.1±3.2 for Supartz/Supartz FX. While both HAs resulted in statistically and clinically meaningful improvements in knee OA symptom severity, there were no observed between-group differences.
Baseline patient characteristics were comparable among all studies ().
Table 2 Study and patient characteristics in randomized controlled trials included in Bayesian meta-analysis
A first analysis included four randomized controlled studies that used WOMAC A as a primary end point, evaluating the efficacy of Supartz/Supartz FX or GenVisc 850 in patients with knee OA. This analysis excluded Karlsson studyCitation18, which was the only one to enroll patients with a Kellgren–Lawrence score of 4. Of the four studies, two were Supartz/Supartz FX vs saline control studies, selected from the five pivotal studies used for regulatory approval, and were included based upon prospective selection criteria that are based upon the availability of WOMAC pain scoresCitation17–Citation19 and two were GenVisc 850 vs saline controlCitation8,Citation20 studies.
Using a Bayesian longitudinal network meta-analysis with a linear trend of studies using WOMAC A as a primary end point and with longer follow-up duration for comparing longitudinally GenVisc 850, Supartz/Supartz FX, and saline control yielded comparable reductions in WOMAC pain scores at 6 weeks, continued reductions in knee pain for >26 weeks in both HA groups, while improvements in knee pain gradually diminished in the saline control group over the same period (). The posterior probability of superiority of GenVisc 850 vs saline control was 79% at 30 weeks, with a 6.9 mm mean advantage (). Additionally, the difference in WOMAC pain scores between the GenVisc 850 and Supartz/Supartz FX remained below the prespecified noninferiority margin of 4 mm throughout the 30-week follow-up period (). Paucity of data toward the end of the time interval causes an increase in variance, and therefore, the posterior probability of noninferiority does not increase. The posterior mean difference between the effect of GenVisc 850 and the reference product was always below the noninferiority margin with a posterior probability of 50%, but the scarcity of data limits the ability to declare noninferiority of GenVisc 850 to the reference product for the interval extending to 30 weeks.
Figure 2 Linear trend in WOMAC pain score posterior mean differences with GenVisc 850, Supartz/Supartz FX, and saline control.
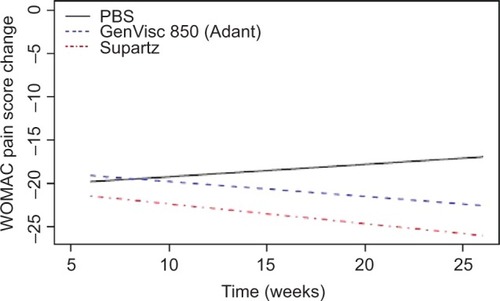
Figure 3 Linear trend in WOMAC pain score posterior mean differences with GenVisc 850, Supartz/Supartz FX, and saline control.
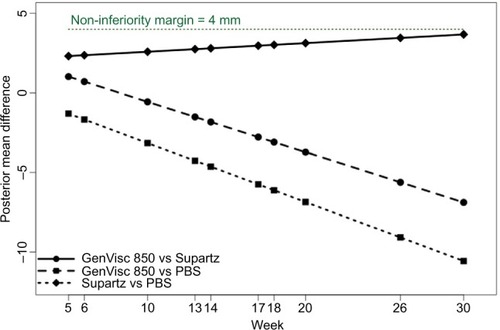
Table 3 WOMAC pain score posterior mean differences with GenVisc 850, Supartz/Supartz FX, and saline control from randomized controlled trials included in Bayesian longitudinal meta-analysis with a linear trend
Efficacy conclusion
Results from two randomized trials comparing Supartz/Supartz FX with saline control, two randomized controlled trials comparing GenVisc 850 with saline, a randomized controlled trial comparing the GenVisc 850 with Supartz/Supartz FX, and a Bayesian meta-analysis integrating efficacy data from published randomized controlled trials all confirm that the clinical efficacy of GenVisc 850 was noninferior to Supartz/Supartz FX and both products were superior to saline control.
Discussion
This report summarizes the clinical evidence that supported a PMA (class III medical device) approval of GenVisc 850 based upon indistinguishable analytical characteristics and noninferior clinical performance to an approved branded product, Supartz/Supartz FX. GenVisc 850 was approved for the treatment of knee OA in September 2015 and is currently marketed in the US, based on indistinguishable physical and chemical characteristics and noninferior clinical performance compared with Supartz/Supartz FX, a previously FDA-approved HA.Citation9
Although the term “generic” is reserved for drugs, and equivalent terminology has been adopted for biologics (biosimilars) and class II medical devices (substantially equivalent), a term has yet to be applied to an approval of a class III medical device. Establishing a defined regulatory pathway for approval of equivalent or indistinguishable devices, specifically for the HA class and class III medical devices, may significantly help to reduce the clinical and economic burden of knee OA. Approximately 18 million adults in the US suffer from symptomatic knee OA, with one in four reporting difficulty with ambulation, despite repeated attempts with traditional nonsurgical therapies.Citation22,Citation23 With the continued aging of the population, the economic burden of knee OA is projected to increase by almost 50% by 2025.Citation23 Recently, intra-articular HA treatment was reported to be among the most effective, long-lasting, and safest treatments for OA knee pain.Citation24–Citation27 Recent projections suggest that the HA market may exceed $1 billion in 2015. Specifically for the reimbursement code for Supartz/Supartz, it is projected that the Center for Medicare and Medicaid Services (CMS) will spend >$700 million over the next 5 years, and it is estimated that GenVisc 850 could save CMS $70–$100 million and patients >$8–$10 million in copayments over this same period through competitive pricing.
Similar to the experience with generic drugs, widespread availability of indistinguishable class III medical devices has the potential to reduce health-care costs while providing equivalent care. However, the first step in establishing such a benefit to the health-care system is that the FDA should establish scientifically valid and rigorous criteria for assessing such equivalence and embrace the legislation formally adopted in 1997 to allow such a process.Citation7 The detailed clinical performance evaluations outlined in this paper and the approval of GenVisc 850 utilizing in part such analysis are the first attempt to validate the analytical methodology to establish such a standard for such evaluations and set the standard for future regulatory reviews.
Importantly, the Bayesian methodology used to support the GenVisc 850 PMA approval is complex and can be more difficult for clinicians and regulatory agencies to understand than the results of a classical frequentist statistical approach to single randomized clinical trials. However, it also has the advantage to consider all available data, as was the case in this report, and the decision to adopt this approach must incorporate a prospectively defined Statistical Analysis Plan. It should be noted that this plan contained more assumptions to be tested (eg, homogeneity over time and exchangeability) and to be verified than might be needed for a randomized clinical trial. The use of this methodology should not be considered the new standard for PMA approval or implemented without advance notice to and acceptance by regulatory agencies.
Conclusion
The preclinical and clinical evidence summarized herein supported FDA approval of a class III medical device based upon indistinguishable characteristics in comparison to an approved branded product. GenVisc 850 was determined to have: 1) noninferior efficacy compared with an FDA-approved HA, 2) superior efficacy vs saline injections, using novel Bayesian modeling, and 3) no unexpected safety risks. Increasing the availability of equivalent or indistinguishable class III medical devices will foster competition and innovation and at the same time significantly reduce health-care spending in the US.
Acknowledgments
The authors thank Doctor David Toledo-Velasquez, PhD, of OrthogenRx, Inc. and Doctor Pilar Coronel-Granado, PhD, for their facilitation in accessing the related data, contributions, and comments.
Disclosure
Doros, Lavin, and Miller were compensated by OrthogenRx, Inc. for their respective role in the conduct of this study. Daley is the President of OrthogenRx, Inc. The authors report no other conflicts of interest in this work.
References
- United States Government Publishing OfficePublic Law 98-417 – September 24, 19841984 Available from: https://www.gpo.gov/fdsys/pkg/STATUTE-98/pdf/STATUTE-98-Pg1585.pdfAccessed September 28, 2015
- IMS Institute for Healthcare Informatics [webpage on the Internet]Medicines Use and Spending Shifts: A Review of the Use of Medicines in the U.S. in 20142014 Available from: http://www.imshealth.com/portal/site/imshealth/menuitem.762a961826aad98f53c753c71ad8c22a/?vgnextoid=3f140a4331e8c410VgnVCM1000000e2e2ca2RCRD&vgnextchannel=736de5fda6370410VgnVCM10000076192ca2RCRD&vgnextfmt=defaultAccessed October 3, 2015
- Generic Pharmaceutical Association [webpage on the Internet]Drug Savings in the US2014 Available from: http://www.gphaonline.org/media/cms/GPhA_Savings_Report.9.10.14_FINAL.pdfAccessed October 3, 2015
- Food and Drug Administration [webpage on the Internet]Biologics Price Competition and Innovation Act of 20092009 Available from: https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=1&cad=rja&uact=8&ved=0CB4QFjAAahUKEwja3a7IjJvIAhXIuB4KHd6hBAQ&url=http%3A%2F%2Fwww.fda.gov%2Fdownloads%2FDrugs%2FGuidanceComplianceRegulatoryInformation%2Fucm216146.pdf&usg=AFQjCNEbnUIJlEgV3lLOj0RZyFtHxK-1OQ&sig2=u7u-QL8P_0AFcvAiEmlvUgAccessed September 28, 2015
- Food and Drug Administration [webpage on the Internet]Filgrastimsndz2015 Available from: http://www.fda.gov/Drugs/InformationOn-Drugs/ApprovedDrugs/ucm436953.htmAccessed September 23, 2015
- Congressional Budget Office [webpage on the Internet]S. 1695, Biologics Price Competition and Innovation Act of 20072008 Available from: https://www.cbo.gov/sites/default/files/110th-congress-2007-2008/costestimate/s16950.pdfAccessed September 29, 2015
- Food and Drug Administration [webpage on the Internet]Guidance for Industry and for FDA Reviewers: Guidance on Section 216 of the Food and Drug Administration Modernization Act of 19972000 Available from: http://www.fda.gov/RegulatoryInformation/Guidances/ucm073707.htmAccessed September 28, 2015
- Navarro-SarabiaFCoronelPCollantesEA 40-month multi-centre, randomised placebo-controlled study to assess the efficacy and carry-over effect of repeated intra-articular injections of hyaluronic acid in knee osteoarthritis: the AMELIA projectAnn Rheum Dis201170111957196221852252
- Food and Drug Administration [webpage on the Internet]Premarket Approval (PMA) – GenVisc 8502015 Available from: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?ID=10038Accessed October 3, 2015
- BraithwaiteGDaleyMJToledo-VasquezDRheological and molecular weight comparisons of approved hyaluronic acid products – preliminary standards for establishing class III medical device equivalenceJ Biomater Sci Polym Ed201627323524626569146
- Food and Drug Administration [webpage on the Internet]Summary of Safety and Effectiveness DataSUPARTZ™ Dispo – P9800442001 Available from: http://www.accessdata.fda.gov/cdrh_docs/pdf/P980044b.pdfAccessed September 29, 2015
- XinYJianhaoLTianshengSThe efficacy and safety of sodium hyaluronate injection (Adant®) in treating degenerative osteoarthritis; a multi-center, randomized, double-blind, positive-drug parallel controlled and non-inferiority clinical studyInt J Rheum Dis201619327127826555862
- CaldwellDMAdesAEHigginsJPSimultaneous comparison of multiple treatments: combining direct and indirect evidenceBMJ2005331752189790016223826
- GouldALUsing prior findings to augment active-controlled trials and trials with small placebo groupsDrug Inf J1991253369380
- LunnDSpiegelhalterDThomasABestNThe BUGS project: evolution, critique and future directionsStat Med200928253049306719630097
- BellamyNBuchananWWGoldsmithCHCampbellJStittLWValidation study of WOMAC: a health status instrument for measuring clinically important patient relevant outcomes to antirheumatic drug therapy in patients with osteoarthritis of the hip or kneeJ Rheumatol19881512183318403068365
- DayRBrooksPConaghanPGPetersenMA double blind, randomized, multicenter, parallel group study of the effectiveness and tolerance of intraarticular hyaluronan in osteoarthritis of the kneeJ Rheumatol200431477578215088306
- KarlssonJSjogrenLSLohmanderLSComparison of two hyaluronan drugs and placebo in patients with knee osteoarthritis. A controlled, randomized, double-blind, parallel-design multicentre studyRheuma-tology (Oxford)2002411112401248
- LohmanderLSDalenNEnglundGIntra-articular hyaluronan injections in the treatment of osteoarthritis of the knee: a randomised, double blind, placebo controlled multicentre trial. Hyaluronan Multi-centre Trial GroupAnn Rheum Dis19965574244318774159
- BlancoFJFernandez-SueiroJLPinto-TasendeJCIntra-articular hyaluronan treatment of patients with knee osteoarthritis waiting for replacement surgeryOpen Arthritis J20081117
- LequesneMGMeryCSamsonMGerardPIndexes of severity for osteoarthritis of the hip and knee. Validation – value in comparison with other assessment testsScand J Rheumatol Suppl19876585893479839
- DillonCFRaschEKGuQHirschRPrevalence of knee osteoarthritis in the United States: arthritis data from the Third National Health and Nutrition Examination Survey 1991-94J Rheumatol200633112271227917013996
- LondonNJMillerLEBlockJEClinical and economic consequences of the treatment gap in knee osteoarthritis managementMed Hypotheses201176688789221440373
- BannuruRRSchmidCHKentDMVaysbrotEEWongJBMcAlindonTEComparative effectiveness of pharmacologic interventions for knee osteoarthritis: a systematic review and network meta-analysisAnn Intern Med20151621465425560713
- StrandVMcIntyreLFBeachWRMillerLEBlockJESafety and efficacy of US-approved viscosupplements for knee osteoarthritis: a systematic review and meta-analysis of randomized, saline-controlled trialsJ Pain Res2015821722826005358
- MillerLEBlockJEUS-approved intra-articular hyaluronic acid injections are safe and effective in patients with knee osteoarthritis: systematic review and meta-analysis of randomized, saline-controlled trialsClin Med Insights Arthritis Musculoskelet Disord20136576324027421
- CampbellKAEricksonBJSaltzmanBMIs local viscosupple-mentation injection clinically superior to other therapies in the treatment of osteoarthritis of the knee: a systematic review of overlapping meta-analysesArthroscopy2015311020362045.e1425998016