
Abstract
Deletions of the 15q26 region encompassing the chromodomain helicase DNA binding domain 2 (CHD2) gene have been associated with intellectual disability, behavioral problems, and several types of epilepsy. Including the cases mentioned in ECARUCA (European cytogeneticists association register of unbalanced chromosome aberrations) and DECIPHER (database of genomic variation and phenotype in humans using ensembl resources), so far, a total of 13 intellectually disabled patients with a genetically proven deletion of the CHD2 gene are described, of whom eleven had a history of severe forms of epilepsy starting from a young age. In this article, a moderately intellectually disabled 15-year-old male with a 15q26.1–q26.2 interstitial deletion is reported, who was referred for analysis of two recent short-lasting psychotic episodes that were nonresponsive to antipsychotic treatment and recurrent disinhibited behaviors since early infancy. Careful interdisciplinary assessment revealed that the psychotic phenomena originated from a previously unrecognized absence epilepsy. Treatment with valproic acid was started which resulted in full remission of psychotic symptoms, and consequently, substantial improvement of behavior. It was concluded that in case of (rare) developmental disorders with genetically proven etiology, a detailed inventory of anamnestic data and description of symptomatology over time may elucidate epilepsy-related psychopathology for which a specific treatment regimen is needed.
Introduction
Microarray analysis has been shown to be of crucial importance for the understanding of the genetic etiology of intellectual disability (ID). With this technique, numerous (de novo) copy number variations have been discovered leading to several circumscribed microdeletion syndromes.Citation1,Citation2 Especially in patients in whom neuropsychiatric disorders or congenital anomalies are present, in addition to ID, extensive genetic analysis is warranted.
Partial deletions of the long arm of chromosome 15, particularly in the 15q26 region, have been typically associated with somatic anomalies, especially congenital diaphragmatic hernia.Citation3 In addition, ID, epilepsy, and behavioral problems are frequently mentioned.Citation4–Citation6 With respect to the 15q26.1 subregion, the chromodomain helicase DNA binding domain 2 (CHD2) gene (OMIM: 602119), originally characterized by Woodage et al,Citation7 has been repeatedly reported to play a pivotal role in cerebrocortical development.Citation8 Both microdeletions encompassing this gene and de novo loss of function mutations lead to a syndrome characterized by ID and epileptic encephalopathy with generalized seizures.Citation9,Citation10
So far, a total of nine patients with ID (seven females and two males) has been reported with a genetically proven deletion of the CHD2 gene. Of these patients, eight had a history of severe forms of epilepsy starting from the young age.Citation4–Citation6,Citation11,Citation12
Here, a 15-year-old male with a 15q26.1–q26.2 interstitial deletion is described, who was referred to the specialized outpatient department for psychopathology and genetics because of short-lasting psychotic episodes and problematic management of disinhibited behaviors for which over many years symptomatic treatment with antipsychotics (ie, haloperidol) was ineffective.
Clinical description
The patient is a 15-year-old male of Chinese origin, who was adopted at the age of 18 months in a malnourished condition from an orphanage by his Dutch foster parents. Shortly after birth, in the People’s Republic of China, he underwent the first surgical correction for bilateral cheilognathopalatoschisis followed by several reconstructive surgeries in the Netherlands up to the age of 5 years. Upon arrival, he was referred to a pediatrician of a university hospital for evaluation of developmental delay as well as bizarre autistic and disinhibited behaviors. At examination, apart from moderate auditory impairment secondary to recurrent upper airway infections, mild motor delay was noticed for which physical therapy was applied. Full laboratory investigation according to the Dutch adoption protocol, including screening for metabolic disorders, disclosed no abnormalities. Electroencephalogram (EEG) registration revealed focal frontocentral abnormalities that were difficult to interpret because of muscular artifacts.
At the age of 2 years, a symptomatic treatment with a low dose of haloperidol was started because of persisting challenging behavior. Subsequently, he was extensively examined by a child neurologist at the age of 5 years. Weight, length, and head circumference were 17.4 kg (−1 standard deviation [SD]), 110 cm (0 SD), and 46 cm (−2 SD), respectively. No abnormalities were found apart from motor overactivity. His attention was undisturbed, whereas communication was adequate with the use of a hearing aid. Since his parents frequently noticed brief moments with a blank staring and unresponsiveness to his surroundings, EEG recording was performed, which showed similar frontal irregularities without, however, specific epileptic configurations. A diagnosis of aspecific psychomotor retardation was made. Six months later, due to aggressive, self-injurious, and autistic behaviors, reevaluation by a pediatrician was performed, who advised a more congruent and structured behavioral approach.
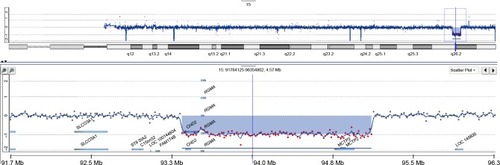
During subsequent years, various interventions with antipsychotics for behavioral control were implemented, which, however, frequently induced unwanted side effects such as dyskinesias, akathisia, and facial edema. In this period, intelligence testing was performed twice with an interval of 3 years showing a total intelligence quotient (IQ) of 55 and a developmental age of ~3.6 years (calendar age: 8 years). At the age of 12 years, microarray analysis (Agilent 180K oligoarray, amadid 023363 [Agilent Technologies, Santa Clara, CA, USA]) demonstrated an 1.8 Mb interstitial deletion in 15q26.1–q26.2, encompassing the CDH2 and RGMA genes that were considered to be etiologically involved (karyotype: arr[hg19]15q26.1q26.2(93,408,532–95,188,862)x1; ). Magnetic resonance imaging scan of the brain showed no abnormalities.
Figure 1 Copy number profile of chromosome 15 of the patient obtained by oligoarray-based comparative genomic hybridization (180K platform).
In the next 18 months, two short-lasting episodes occurred characterized by marked global regression, anxieties, hallucinatory experiences (visual and auditory), as well as disinhibited and aggressive behaviors. Treatment with haloperidol and promethazine in a twice-daily dose of 1.5 and 25 mg, respectively, was prescribed with, however, insufficient efficacy. As a consequence, the patient was referred to the specialized outpatient facility for psychopathology and genetics for integrative assessment of behavior and psychopathology in the context of the genetic condition. Written informed consent has been obtained from the mother of the patient. This study has been approved by the Vincent van Gogh Institutional Review Board.
Results
At examination, a young male adolescent was seen with a status after multiple reconstructive surgical corrections for bilateral cheilognathopalatoschisis. No dysmorphias were noticed. His behavioral repertoire comprised compulsive elements, hyperactivity, and impulsivity. No major psychiatric symptoms could be identified. Detailed heteroanamnestic evaluation revealed that, since the age of 13 years, two short-lasting psychotic episodes had occurred dominated by anxieties, dysphoric mood swings, threatening and disinhibited behaviors, physical aggression, and both visual and auditory hallucinations, accompanied by diminished consciousness as well as restricted interpersonal contact.
Routine hematological and biochemical laboratory tests were all normal. Pharmacogenetic analysis demonstrated no abnormalities for CYP2C9 and CYP2C19. For CYP2D6, however, a *10/*10 genotype was found that is in accordance with a significantly impaired biotransformation capacity.
Extensive neuropsychological assessment (Vineland screener,Citation13 VISKCitation14) showed a developmental age of 2 years with a discrepant profile (social: 0.8 years; motor and communication: >2.6 years). Formal intelligence testing could not be fully performed due to major impairments of attention and inhibitory functioning as well as repetitive behaviors. The latter was objectivated by proxy ratings of attention and executive functioning (BRIEF,Citation15 BADS-DEXCitation16). Assessment of psychopathology by means of the psychopathology instrument for mentally retarded adults pointed specifically at chaotic, anxious, and generally inadaptive behaviors (total score: 32; maximum: 56).Citation17
Based on the aforementioned information, a preliminary diagnosis of epilepsy-related recurrent brief psychosis was made and it was advised to perform EEG recording. The latter revealed that some epileptic paroxysms left frontotemporal and sporadic polyspike–wave complexes. For this reason, the antiepileptic compound valproic acid was prescribed, whereas symptomatic treatment for behavioral control with a low dose of haloperidol (1.6 mg daily [gtt]; plasma concentration: 1 μg/L) was continued. This antiepileptic intervention (600 mg valproic acid; plasma concentration: 85 mg/L) resulted in a full remission of psychotic symptoms and a gradual normalization of behavior to the premorbid level that, apart from incidentally occurring odd beliefs, was paralleled by stabilization of mood and marked improvement of social adaptation.
Discussion
In the moderately intellectually disabled patient presented here, psychotic phenomena appeared to be the result of a previously unrecognized absence epilepsy. Treatment with valproic acid induced a substantial improvement of his behavior while keeping free from further psychotic episodes. As stressed by several authors, epilepsy-related psychoses, both post- and interictal, are still a clinical challenge to recognize and treat appropriately.Citation18,Citation19
The 15q26.1 microdeletion, encompassing among others the CHD2 gene, is in general associated with febrile seizures starting at early age followed by various epileptic features, myoclonic as well as tonic–clonic and absence seizures, and not typically clear dysmorphisms.Citation9,Citation10 In the current patient, absence epilepsy was only present. Out of a total of nine published cases, this type of epilepsy is mentioned in two, whereas in one, no epilepsy was present (). As can be inferred, in addition to the published cases, four patients are registered in international databases of whom one (DECIPHER 304608) was not diagnosed with epilepsy. Finally, two patients were detected in large copy-number variation studies, but no clinical information about their phenotype or presence of epilepsy is provided.
Table 1 Reported patients with a 15q26 microdeletion encompassing the CHD2 gene, intellectual disability, and challenging behaviorsTable Footnotea
In eight patients, the 15q26.1 deletion also encompassed the RGMA gene (OMIM: 607362) that is assumed to be involved in immune functionality but not regarded to be of clinical significance for ID or epilepsy.Citation20
Conclusion
This case demonstrates the vital importance of detailed anamnesis and careful description of the symptom–cognition–behavior complex over time, both to ensure early detection of epilepsy-related psychopathology, especially in people with ID, and to avoid diagnostic misinterpretation and inappropriate treatment.
Disclosure
The authors report no conflicts of interests in this work.
References
- VissersLEde LigtJGilissenCA de novo paradigm for mental retardationNat Genet2010421109111321076407
- GilissenCHehir-KwaJYTjwan ThungDGenome sequencing identifies major causes of severe intellectual disabilityNature201451134434724896178
- KlaassensMGaljaardRJScottDAPrenatal detection and outcome of congenital diaphragmatic hernia (CDH) associated with deletion of chromosome 15q26: two patients and review of the literatureAm J Med Genet2007143A22012212
- VerediceCBiancoFContaldoIEarly onset myoclonic epilepsy and 15q26 microdeletion: observation of the first caseEpilepsia2009501810181519486360
- DhamijaRBreningstallGWong-KisielLDolanMHirschBWirrellEMicrodeletion of chromosome 15q26.1 in a child with intractable generalized epilepsyPediatr Neurol201145606221723464
- CapelliLPKrepischiACVGurgel-GiannettiJDeletion of the RMGA and CDH2 genes in a child with epilepsy and mental deficiencyEur J Med Genet20125513213422178256
- WoodageTBasraiMABaxevanisADHieterPCollinsFSCharacterization of the CDH family of proteinsProc Natl Acad Sci U S A19979411472114779326634
- ShenTJiFYuanZJiaoJCHD2 is required for embryonic neurogenesis in the developing cerebral cortexStem Cells2015331794180625786798
- SulsAJaehnJAKecskésADe novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndromeAm J Hum Genet20139396797524207121
- CarvillGLHeavinSBYendleSCTargeted resequencing in epileptic encephalopathies identifies de novo mutation in CHD2 and SYNGAP1Nat Genet20134582583023708187
- CourageCHougeGGallatiSSchjelderupJRieublandC15q26.1 microdeletion encompaasing only CHD2 and RGMA in two adults with moderate intellectual disability, epilepsy and truncal obesityEur J Med Genet20145752052324932903
- ChénierSYoonGArgiropoulosBCHD2 haploinsufficienty is associated with developmental delay, intellectual disability, epilepsy and neurobehavioural problemsJ Neurodev Disord20146924834135
- ScholteEVan DuijnEDijkxhoornYVineland Screener 0–6 Years: Manual of the Dutch AdaptationLeidenPITS2008
- HartmanCLuteijnEMoorlag-JonkerHde BildtAMinderaaRVISK Test ManualAmsterdamBoom2015
- GioiaGIsquithPGuySKenworthyLBehavior Rating Inventory of Executive Functioning (BRIEF): Professional ManualLutz, FLPsychological Assessment Resources2000
- WilsonBAldermanNBurgessPEmslieHEvansJBADS: Behavioural Assessment of the Dysexecutive SyndromeLondonPearson1997
- MatsonJLKazdinAESenatoreVPsychometric properties of the psychopathology instrument for mentally retarded adultsAppl Res Ment Retard1994581896721483
- MaramatsuRKuboTMoriMRGMa modulates T cell responses and is involved in autoimmune encephalomyelitisNat Med20111748849421423182
- GaitatzisATrimbleMRSanderJWThe psychiatric comorbidity of epilepsyActa Neurol Scand200411020722015355484
- VerhoevenWMAEggerJIMGunningWBBeversMde PontBJRecurrent schizophrenia-like psychosis as first manifestation of epilepsy: a diagnostic challenge in neuropsychiatryNeuropsychiatr Dis Treat2010622723120520786
- PintoDPagnamentaATKleiLFunctional impact of global rare copy number variation in autism spectrum disorderNature201046636837220531469
- HamdanFFSroerMCapo-ChichiJMDe novo mutations in moderate or severe intellectual disabilityPLoS Genet201410e100477225356899