Abstract
At present, more than 100 disease mutations in mitochondrial DNA polymerase γ (POLG) have been indentified that are causally related to an array of neuropsychiatric diseases affecting multiple systems. Both autosomal recessive and autosomal dominant forms can be delineated, the latter being associated with Parkinsonism and depressive or psychotic syndromes. In this report, a middle-aged female patient with recurrent major depression with melancholic features, slowly progressive gait instability, and dilated cardiomyopathy is described. Detailed diagnostic evaluation was performed to elucidate the supposed relationship between ataxia, cardiomyopathy, and major depression with melancholia. After extensive genetic and metabolic investigation, a nucleotide substitution c.2207 A→G in the POLG gene resulting in amino acid change Asn 736Ser in exon 13 was demonstrated. This mutation was considered to be compatible with a mitochondrial disorder and implicated in the pathophysiology of the neuropsychiatric syndrome. It is concluded that this novel POLG mutation forms the most parsimonious etiological explanation for the here-described combination of ataxia, major depression, and cardiomyopathy. Therefore, in patients with a complex neuropsychiatric presentation, extensive diagnostic analysis is warranted, including the search for mitochondriopathies, in order to avoid unnecessary delay of adequate treatment.
Introduction
Cerebellar ataxias, of which the spinocerebellar ataxias (SCAs) are the most prevalent, represent a heterogeneous group of neurodegenerative disorders that, according to their genetic etiology, can be differentiated among autosomal dominant, autosomal recessive, X-linked, and maternally inherited syndromes.Citation1,Citation2 Over the last decade, novel mitochondrial genetic diseases have been identified in which mutations in DNA polymerase γ (POLG [MIM 174763]) gene are involved. POLG1 is the only DNA polymerase in human mitochondria and is essential for mitochondrial (mt)DNA replication and repair. It has to be stressed that functional genetic variants of POLG are present in about 0.5 percent of the normal population. Defects in mtDNA replication lead to mitochondrial dysfunction and disease. Originally, primary mtDNA mutations were thought to be the major cause of human mitochondriopathies.Citation3 Nowadays, however, a great number of novel diseases have been described in which POLG mutations are causative through a secondary effect on mtDNA. These syndromes are thought to have a common European ancestry and may manifest from early infancy to late middle age.Citation4,Citation5
To date, POLG mutations have been found to be causal in a great number of mitochondrial diseases with a very heterogeneous clinical manifestation and affecting multiple systems.Citation6 In 2001, Van Goethem and coworkers identified a mutation in POLG in a Belgian pedigree with progressive external ophthalmoplegia.Citation7 Thereafter, mutations in POLG were found in a great variety of diseases like Alpers syndrome, SCA negative cerebellar ataxia, and Charcot-Marie-Tooth disease.Citation8–Citation10
POLG mutations cause an overlapping clinical spectrum of diseases with both dominant and recessive modes of inheritance. Autosomal recessive forms often present with external ophthalmoplegia, peripheral neuropathy, ataxia, and epilepsy.Citation5,Citation11 Autosomal dominant forms are mostly characterized by symptoms within the ataxia-neuropathy range and by progressive external ophthalmoplegia. In addition, these autosomal dominant syndromes are reported to be associated with an array of neuropsychiatric symptoms from the Parkinson spectrum and from the psychotic and depressive domains of which major depression has the highest incidence.Citation12–Citation14
In this report, a middle-aged female patient is described with a long history of recurrent depression and slowly progressive gait instability, as well as dilated cardiomyopathy for the 5-year period prior to admission. A descriptive diagnosis of progressive cerebellar ataxia with bilateral pyramidal syndrome and cardiomyopathy was made. Later, extensive genetic work-up demonstrated a POLG mutation which was thought to be of pathogenic significance.
Clinical observations and results
The patient is a 52-year-old single female born from non-consanguineous parents. She has three healthy sisters. Her only brother died in his early fifties, allegedly from gastric hemorrhage. Her mother and father died aged 55 and 80 from acute cardiac arrest, most probably due to cardiomyopathy, and cerebral hemorrhagia respectively. There is no family history of mental disorders or hereditary diseases. At the age of 33 she gave birth to a healthy son who is mildly mentally retarded. The patient showed a normal developmental trajectory, completed primary school followed by domestic training, and was subsequently employed in nondemanding jobs. Aged 17, she was involved in a serious car accident resulting in cerebral concussion and various fractures of the right foot and knee for which she underwent several orthopedic surgical corrections. Since that time, the patient showed an unstable gait and balance, which with physical therapy gradually disappeared. For a period of 20 years, she was able to perform her elementary activities at a laundry, not hindered by motor impairments. At the age of 37 years, however, she was referred to a rehabilitation physician because she’d had slowly progressive gait instability for a period of 2 years. Physical examination disclosed a mild ataxia that was interpreted as a result of the cerebral concussion. In addition, emotional and affective instability were noticed. No neuroradiological investigation was performed.
Over subsequent years, the patient showed recurrent depressive symptoms for which she was treated by her general practitioner with several antidepressants. Aged 44, she underwent surgery for bilateral subcapsular posterior cataract. At the same age, she was admitted to a psychiatric hospital because of domestic neglect, inactivity, paranoid ideation, and symptoms of major depression with melancholic features. Neurological examination demonstrated bilateral ataxia and a slightly dysarthric speech. CT (computed tomography)-scanning of the brain revealed a moderate cerebellar atrophy. The neurological symptoms were considered to be of posttraumatic origin. Treatment with clomipramine resulted in complete remission of depressive symptoms, and the patient was followed up at the outpatient department of psychiatry. At the age of 47, she developed complaints of fatigue and shortness of breath for which she was referred to a cardiologist. A diagnosis of dilated cardiomyopathy with a cardiac output of less than 30% was made, and treatment with the beta blocker bisoprolol was started. Over the subsequent 6 years, several depressive episodes with psychotic phenomena and suicidal events occurred that were treated with various psychotropic agents. Because of serious impairment of general functioning and persistent depressive and psychotic symptoms, the patient was referred to the specialized department of neuropsychiatry for extensive diagnostic assessment.
At admission, extreme loss of energy and marked hypokinesia were prominent, and as a consequence, the patient was partially wheelchair bound. Since these phenomena were caused by her cardiomyopathy and use of antipsychotics respectively, treatment with bisoprolol was first optimalized and psychotropics were gradually tapered off. Thereafter, neuropsychiatric examination revealed a slight paranoid ideation and suicidal thoughts. There was significant loss of initiative, flattening of affect, depressed/dysphoric mood, and delusional ideas of nihilism, as well as anosognosia. Neuropsychological functioning was characterized by markedly lowered information processing and a relatively intact working memory. Her total IQ was 69 (WAIS-IIICitation15), indicative of below average intellectual functioning. The patient was friendly and cooperative but refused an extensive testing program. ECG-recording showed an incomplete left ventricular bundle branch block with inverted T-waves. Routine hematological and biochemical laboratory tests disclosed a mild dyslipidemia only (cholesterol 6.7 mmol/L, with other lipid parameters normal).
Treatment with citalopram in a maximal dose of 20 mg daily resulted in a gradual reduction of depressive symptoms, even though inactivity and loss of energy persisted and cerebellar dysfunctions became more prominent. An MRI (magnetic resonance imaging) scan of the brain demonstrated marked cerebellar atrophy without any supratentorial lesions (). Karyotyping revealed no abnormalities.
Figure 1 Axial, T2-weighted magnetic resonance image of the patient, showing atrophy of the cerebellum and the cerebellar vermis.
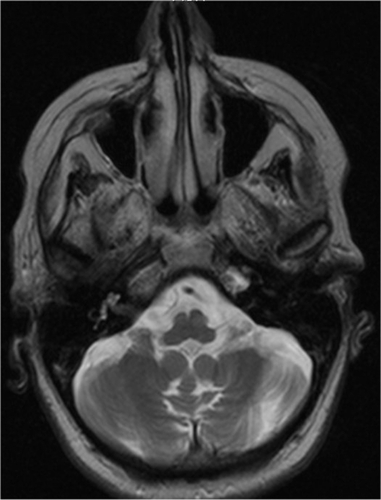
Given the combination of ataxia and cardiomyopathy, the possibility of a hereditary spinocerebellar ataxia was considered, and hence an extensive neurological and etiological diagnostic procedure was started.
At neurological examination, the patient presented with slow speed and lack of initiative. Funduscopy revealed no abnormalities. There were saccadic intrusions of ocular smooth pursuit. Minimal bilateral thenar atrophia was noticed. No pareses were present. Speech was dysarthric and gait slightly ataxic but with normal base. All cerebellar tests were impaired. Sensibility was undisturbed in all modalities. Tendon reflexes of the arms and legs were hyperactive with bilateral positive Hoffmann–Trömner and Babinski signs. Snout reflex was present. An initial descriptive diagnosis of progressive cerebellar ataxia with bilateral pyramidal syndrome and cardiomyopathy, present for over a decade, was given. With mutation analysis, all known autosomal recessive cerebellar ataxias were excluded. Metabolic diseases like cerebrotendinous xanthomatosis and sphingolipidoses, especially Niemann–Pick disease type C, could not be demonstrated. The spectrum of amino acids in plasma as well as the levels of lactate and pyruvate were all normal. Mutation analysis of the POLG gene demonstrated a nucleotide substitution c.2207 A→G resulting in amino acid change Asn 736Ser, which with sequence analysis appeared to be located in exon 13. This mutation was considered to be compatible with a mitochondrial disorder and assumed to be implicated in the pathophysiology of the neurological syndrome and the cardiomyopathy.
Thus, a provisional diagnosis of a mitochondrial POLG disease was made, probably an autosomal dominant heterozygotic form. Unfortunately, its clinical significance could not further be substantiated, since family members, in particular the son, were not available for additional testing. Although the depressive symptoms were no longer prominent, chronic hospitalization was necessary because of a slowly progressive increase of both cardiac dysfunction and neurological symptoms.
Discussion
The case described here is a typical example of a patient in whom an apparently common psychiatric disease, major depression, was initially diagnosed. Detailed recording of her medical life chart and family history, however, was indicative of the presence of a complex neurodegenerative disorder characterized by slowly progressive cerebellar ataxia and cardiomyopathy. Such a combination of symptoms goes along with a vast amount of differential diagnostic options, including hereditary ataxias and metabolic disorders (eg, lysosomal storage diseases, disturbances in amino acid metabolism, and cerebral cholesterosis). By means of extensive laboratory and genetic testing, all diagnostic hypotheses, other than a POLG mutation disease, could be falsified. In this patient, therefore, a tentative diagnosis of a POLG mis-sense mutation was made.
Since there is an abundance of nucleotide substitutions that have been described in patients, Horvath and colleagues point at the diagnostic challenge that has to be mastered and provide a set of heuristic decision rules that delimit the clinical significance of a POLG mutation.Citation16 The first principle states that the amino acid change is located in a functional relevant site of POLG. A second criterion is that that the mutation under study should be close to other established pathogenic POLG mutations. Given these principles and, in this case, the constellation of symptoms and the nucleotide change in the mutation c.2207 A→G, this novel mutation was considered to be pathogenically involved despite there being no information available on this mutation in normal populations.
It is well known that disorders within the mood and anxiety domain may be the initial sign of mitochondrial disorders. In fact, the prevalence in mitochondriopathies of a lifetime diagnosis of major depressive disorder is 54%.Citation17 From several studies, it emerges that impairment of metabolism, especially at the mitochondrial level, may be involved in the pathophysiology of psychiatric diseases, including depressive disorders.Citation18 Also POLG mutations have been reported to be associated with major depression.Citation13 Therefore, major depression with melancholia as present in the current patient may be considered as part of the POLG-associated neuropsychiatric syndrome.
In conclusion, the presented case demonstrates a previously unreported POLG mutation accompanied by ataxia, cardiomyopathy, and major depression. Given the very heterogeneous phenotypical presentation of this multisystem disease, mutations involving the POLG gene should be considered in patients with major depressive disorder in whom a combination of unexplained neurological (eg, ataxia) and nonneurological (eg, cardiac dysfunction) is present.
Acknowledgements
This study is part of a collaborative project of the research group ‘Psychopathology and Genetics’.
Disclosure
The authors report no conflicts of interest in this work.
References
- CopelandWCInherited mitochondrial diseases of DNA replicationAnn Rev Med20085913114617892433
- FinstererJAtaxias with autosomal, X-chromosomal or maternal inheritanceCan J Neurol Sci20093640942819650351
- FinstererJMitochondriopathiesEur J Neurol20041116318615009163
- HakonenAHDavidzonGDalemiRAbundance of the POLG disease mutations in Europe, Australia, New Zealand, and the United States explained by single ancient European foundersEur J Hum Genet20071577978317426723
- HudsonGChinneryPFMitochondrial DNA polymerase-γ and human diseaseHum Mol Genet200615R244R25216987890
- HakonenAHHeiskanenSJuvonenVAm J Hum Genet20057743044116080118
- Van GoethemGDermautBLöfgrenAMutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletionsNat Genet20012821121211431686
- ChanSSLCopelandWCDNA polymerase gamma and mitochondrial disease: understanding the consequences of POLG mutationsBiochim Biophys Acta2009178731231919010300
- BlokMJvan den BoschBJJongenEThe unfolding clinical spectrum of POLG mutationsJ Med Genet20094677678519578034
- WongLJCNaviauxRKBrunetti-PierriNMolecular and clinical genetics of mitochondrial diseases due to POLG mutationsHum Mutat201029E150E17218546365
- WintherthunSFerrariLHeLAutosomal recessive mitochondrial ataxic syndrome due to mitochondrial polymerase-γ mutationsNeurology2005641204120815824347
- LuomaPMelbergARinneJOParkinsonism, premature menopause, and mitochondrial DNA polymerase-γ mutations: clinical and molecular genetic studyLancet200436487588215351195
- KoeneSKoziczTLRodenburgRJTMajor depression in adolescent children consecutively diagnosed with mitochondrial disorderJ Affect Disord200911432733218692904
- JouSHChiuNYLiuCSMitochondrial dysfunction and psychiatric disordersChang Gung Med J20093237037919664343
- WechslerDWAIS-III Technical ManualSan Antonio, TXPsychological Corporation2002
- HorvathRHudsonGFerrariGPhenotypic spectrum associated with mutations of the mitochondrial polymerase-γ geneBrain20061291674168416621917
- FattalOLinkJQuinnKPsychiatric comorbidity in 36 adults with mitochondrial cytopathiesCNS Spectr20071242943817545953
- RezinGTAmboniGZugnoAIMitochondrial dysfunction and psychiatric disordersNeurochem Res2009341021102918979198