
Abstract
Alzheimer’s disease (AD) is the most common neurodegenerative disease. Histological characterization of amyloid plaques and neurofibrillary tangles in the brains of AD patients, alongside genetic studies in individuals suffering the familial form of the disease, has fueled the accumulation of the amyloid-β protein as the initial pathological trigger of disease. Association studies have recently showed that cerebral hypoxia, via both genetic and epigenetic mechanisms, increase amyloid-β deposition by altering expression levels of enzymes involved in the production/degradation of the protein. Furthermore, hypoxia has also been linked to neuronal and glial-cell calcium dysregulation through formation of calcium-permeable pores, dysregulated glutamate signaling, and intracellular calcium-store dysfunction. Hypoxia has also been strongly linked to neuroinflammation; however, this relationship to AD has not been thoroughly discussed in the literature. Here, we highlight and organize critical research evidence showing that in both hypoxic and AD brains, there are similarities in terms of 1) the substances mediating/modulating the neuroinflammatory environment and 2) the immune cells that drive the formation of these substances.
Introduction
The Medical Research Council defines neurodegenerative diseases as “incurable conditions in which nerve cells gradually degenerate or die”.Citation1 Dementias are a group of neurodegenerative diseases characterized by symptoms including memory loss and difficulties in thinking, problem-solving, or language. In the UK, dementia affects around 850,000 individuals, with an average economic impact of £26 billion a year. This figure is forecast to reach >1 million by 2025 and >2 million by 2051.Citation1 Dementia commonly affects the elderly. In the UK, >7% of persons aged >65 years have dementia.Citation1 Worldwide, 50 million patients have dementia, with a global economic burden of US$818 billion, which is estimated to triple by 2050.Citation2 Therefore, dementia has been highlighted as a growing global health problem that needs to be tackled.
Alzheimer’s disease (AD) accounts for 60%–70% of all dementia cases.Citation3 It is a multifactorial disease caused by both genetic and environmental factors that contribute to and accelerate the progression of the disease. Less than 5% of all AD cases are due to genetic mutations giving rise to early-onset familial AD. The vast majority of AD cases are sporadic, and there is no defined cause; however, risk factors include aging, chronic inflammation, cerebrovascular disease, diabetes mellitus, and stroke. In particular, cerebral hypoxia has been strongly associated with increased risk of developing AD. Hypoxia, the lack of oxygen, is one detrimental aspect of ischemia. This is when tissue is underperfused with blood, and in the brain this can cause hypoxia, impaired delivery of metabolic fuels, and accumulation of metabolites in the brain parenchyma. As such, it is difficult to conclude from studies involving stroke patients or ischemic animal models whether the results are solely due to hypoxia or other downstream effects of ischemia. One longitudinal study found the incidence of dementia increased by approximately sixfold in ischemic stroke patients when compared to controls.Citation2 Furthermore, they showed that in the ischemic stroke patient group, those that suffered other comorbidities that cause cerebral hypoxia had an even higher incidence of AD. Studies showing this have driven research in recent decades into the molecular and cellular links between hypoxia and AD. Here, we review prevailing well-documented links between hypoxia (both ischemia-dependent and -independent) and AD, such as amyloid-β (Aβ) accumulation and calcium-homeostasis dysregulation, and also review the effects of hypoxia on neuroinflammation and its relationship to AD, a link that has not been adequately reviewed in the literature.
Aβ accumulation
In 1907, Aloid Alzheimer produced the first case report of a patient suffering psychiatric illness associated with specific histomorphological changes, such as amyloid plaques (APs) and neurofibrillary tangles (NFTs), which are now widely accepted as the key pathological hallmarks of AD.Citation3 From these hallmarks, two main hypotheses regarding the initial pathological trigger of AD were considered and are discussed herein: the amyloid-cascade hypothesis (ACH) and the tau hypothesis.
It was the identification of the Aβ protein in APs alongside the identification of genetic mutations in APP in sufferers of early-onset familial dementia that resulted in the ACH.Citation4 According to this hypothesis, the deposition and accumulation of Aβ monomers into toxic oligomers is the initial pathological trigger of the disease, which subsequently leads to the formation of NFTs, neuronal cell death, and dementia. This hypothesis has continued to gain support for 25 years. An important breakthrough in the theory was the identification of genetic mutations in subunits of an enzyme (PS1 and PS2 in the y-secretase enzyme) that alter APP processing to promote Aβ formation in suffers of early-onset AD.
The tau hypothesis has also been proposed to explain the initial pathological trigger of AD. Tau proteins bind to and stabilize microtubules and are abundant in neurons of the central nervous system (CNS). The tau hypothesis states that excessive or abnormal phosphorylation of tau results in its transformation into paired helical filaments and NFTs, leading to neuronal death and dementia. This hypothesis is supported by the fact that mutations in the tau gene have been linked to frontotemporal dementia;Citation5 however, mutations in tau have not yet been directly linked to AD.
Although there is evidence for both hypotheses in the initiation of AD pathology, since a mutation in tau gives rise to NFTs but not APs, yet mutations in APP, PS1, or PS2 give rise to both APs and NFTs suggests that Aβ deposition and accumulation occurs upstream of tau phosphorylation.Citation6 Therefore, here we focus on the ACH of Aβ accumulation being the initial pathological trigger of AD.
Hypoxia drives Aβ accumulation
Hypoxia modulates APP metabolism, leading to increased formation of Aβ via the amyloidogenic pathway. Time-dependent hypoxic upregulation of APP has been well documented at both the mRNACitation7 and protein level, following 10–180 mins of ischemia.Citation8 In 1997, Mattson et al proposed that this may act as a defense mechanism in an attempt to increase levels of neuroprotective soluble APPα.Citation9 However, in many cases the increased APP fuels increased levels of Aβ, not soluble APPα.Citation7,Citation8 This is most likely due to hypoxia favoring APP metabolism via the amyloidogenic pathway.
Chronic hypoxia decreases protein levels of ADAM10, a putative α-secretase, which thereby decreases APP cleavage via the nonamyloidogenic pathway.Citation10 This may be a posttranslation effect, as studies have shown that hypoxia decreases the mature form of ADAM10 and increases the immature form, consequently reducing α-secretase processing of APP.Citation11 However, ADAM17, a related sheddase that also processes APP and TNFα, shows decreased gene-expression levels following 3 days of chronic hypoxia.Citation12
In addition, studies have shown that chronic hypoxia increases expression of β-secretase (BACE1), which promotes the amyloidogenic pathway.Citation13 This occurs in both in vitro (cell lines kept at 8% oxygen for 16 hours a day for 1 month) and in animal-stroke models.Citation14 The discovery of hypoxic-response elements in the promoter of BACE1, where the transcriptional factor HIF1α can bind during hypoxia (while it is degraded under normal conditions), alongside the discovery that HIF1α-deficient mice reduce BACE1 expression, pointing to HIF1α as the key mediator of hypoxic induction of BACE1.Citation13 As well as hypoxic stabilization of HIF1α, HIF1α is also upregulated by Aβ.Citation13 Therefore, this introduces a positive-feedback loop, leading to increasing BACE1 levels and other HIF1-induced genes that increase Aβ production, such as (APH1, one component of the γ-secretase complex involved in proteolysis.
γ-Secretase is a complex of presenilin (PS), PEN2, APH1, and Nct. Hypoxia has been shown to increase expression of some of these subunits of γ-secretase. For example, hypoxia-mediated increase in the expression of the APH1 subunit may be due to binding of HIF1α at the hypoxic response–element motif at the promoter region of the APH1 gene.Citation15 Moreover, hypoxia-mediated increases in PS1 levels have been reported at both the mRNA and protein levelCitation16 and in both in vitro and in ischemic animal models.Citation14 Evidence has shown that this upregulation, and the upregulation of PEN2 and NC2, may relate to an epigenetic modification.Citation17 Liu et al showed that chronic hypoxia decreased expression of DNMT3β, which led to demethylation in the CpG site PEN2 and the Nct and PS1 gene promoters. This led to transcriptional activation, resulting in increased PEN2, Nct, and PS1 protein levels, thereby increasing γ-secretase activity. From these results, the group further hypothesized that hypoxia-mediated DNMT inhibition could be a possible mechanistic explanation for increased levels of APP and both β-secretase and γ-secretase.
Hypoxia reduces expression of enzymes that break down Aβ, in particular the metalloprotease neprilysin (NEP).Citation18 In one study, these cells were incubated for 24 hours in <1% oxygen. This may be due to hypoxic upregulation of the GPa histone methyltransferase and HDAC1 through experimental inhibition of HDAC1 by valproic acid, causing decreased Aβ42 deposition in mice.Citation19 Another mechanism involves hypoxic induction of caspases. The APP intracellular domain (AICD), a product of the amyloidogenic pathway, has been shown to bind to the promoter and activate transcription of the NEP gene. This may act as a protective mechanism to reduce the effects of neurotoxic Aβ. However, AICD is also a substrate of caspases, and so hypoxic induction of caspases may lead to increased cleavage of AICD, thereby reducing NEP levels.Citation18 Also, caspase 3 has also been shown to cleave PS1/2, thus altering the cleavage activity of γ-secretase. Therefore, an alternative mechanism is that γ-secretase cleavage of APP leads to the formation of a C-terminal fragment of APP, which (unlike AICD) is incapable of translocating to the nucleus and/or regulating the transcription of NEP.Citation18 The mechanism may also apply to another enzyme that breaks down Aβ, TTR, since there is evidence that TTR is both AICD-regulated and responsive to hypoxia.Citation18
Aβ accumulation and hypoxia cause dysregulation of Ca2+ homeostasis
Neuronal death and loss of synaptic integrity in particular areas of the brain lead to the pathology seen in AD.Citation20 As previously discussed, Aβ accumulation is widely considered the initial pathological trigger of AD; however, the process by which it leads to neuronal death is not fully understood. Many papers have suggested a role of dysregulation of calcium homeostasis. This “calcium hypothesis of AD” was proposed after observation of calcium-signaling dysregulation in animal models of familial AD and postmortem studies of sporadic AD.Citation21 Calcium is an important intracellular ion that regulates many physiological processes in neurons, such as neurotransmitter release, second-messenger signaling, and neuronal excitability. There is an overall inward electrochemical gradient for calcium entry after plasma-membrane calcium-channel opening, but in healthy neurons this intracellular calcium increase is quickly restored to homeostatic levels, due to efflux pumps (specifically the sodium–calcium exchanger) and buffering by the endoplasmic reticulum (ER) and mitochondria. However, high levels of intracellular calcium can trigger cell death (neurotoxicity), due to dysregulation of calcium homeostasis. This mechanism is complex, but one important aspect is mitochondrial calcium overload, leading to leakage of mitochondrial membranes. This increased mitochondrial permeability causes release of reactive oxygen species (ROS) and proapoptotic proteins (such as cytochrome C), which ultimately results in caspase activation and apoptosis.
Aβ accumulation has been linked to calcium overload in neuronal cells. One mechanism for this is Aβ forming calcium-conducting pores in the plasma membrane, leading to calcium influx.Citation22 A second mechanism involves Aβ impairment of NMDA receptors (glutamate-sensitive calcium-permeable channels) via induction of membrane-lipid peroxidation.Citation23 Moreover, Aβ accumulation has not only been linked to calcium-homeostasis dysfunction but also to mediators of apoptosis downstream of the calcium dysfunction. For example, Aβ accumulation has been linked to neuronal cell death via its ability to 1) induce mitochondrial dysfunction,Citation24 2) increase oxidative stress, thus leading to ROS-mediated activation of ASK1, which activates JNK and triggers apoptosis,Citation25 and 3) induce caspase 2 activity.Citation26
Furthermore, astrocyte calcium dyshomeostasis has been linked to neuronal cell death.Citation27 Astrocytes are glial cells found in the CNS, and one of its functions, among many, is to provide support to local neuronal cells.Citation28 Some of these support mechanisms include supplying neurons with antioxidant glutathione (thus reducing neuronal ROS formation) and removal of excess extracellular glutamate. Dysregulation of calcium homeostasis in these astrocytes has been reported to impair this support function, thus leading to increased ROS production and glutamate signaling, events that can ultimately result in neuronal cell death. Aβ has been shown to disrupt calcium homeostasis in astrocytes via formation of calcium-conducting pores.Citation27
Hypoxia can lead to calcium-homeostasis dysregulation through promoting Aβ accumulation. This was initially shown in PC12 cells,Citation29 where cells were exposed to 5% oxygen for 24 hours before experimentation. It was later reproduced in primary cultures of central neurons exposed to 2.5% oxygen for 24 hours,Citation30 where hypoxia led to increased intracellular calcium concentrations by upregulation of L-type calcium channels, an effect that was reproduced upon application of exogenous Aβ accumulation under normoxic conditions. Addition of inhibitors of β- or γ-secretase prevented this hypoxic augmentation of calcium currents, thus demonstrating that this effect of hypoxia was dependent on the presence of Aβ.Citation29,Citation30 This ability of Aβ to alter trafficking of the channel may relate to the ability of Aβ to interact closely with the α-subunit of the L-type channel.Citation31
Hypoxia is also able to result in calcium dyshomeostasis in neurons independently of Aβ accumulation by acting on astrocyte physiology. Cells lines maintained with 1% oxygen for 24 hours led to suppression of glutamate-uptake transporters found on astrocytes,Citation32 which results in increased glutamate in extracellular fluid and excitotoxic neuronal death. The mechanism for this neuronal death was originally believed to be increased influx of sodium and chloride ions through glutamate-sensitive AMPA receptors, leading to water entry and swelling of the neurons.Citation33 However, this swelling was usually reversible, and an alternative mechanism for delayed cell death may be extracellular glutamate acting directly via NMDA and AMPA receptors or indirectly via metabotropic glutamate receptors to increase calcium influx into neurons.Citation33
Furthermore, hypoxia has also been shown to dysregulate calcium homeostasis directly in astrocytes (which in turn leads to neuronal death), once again independently of Aβ accumulation. In addition, intracellular calcium levels after addition of bradykinin (an agonist of the BK2 G-protein-coupled receptor that mobilizes calcium from the ER) was augmented in hypoxic astrocytes.Citation34 The mechanism proposed was that hypoxia led to hyperpolarization of the mitochondria, increasing the mitochondria’s calcium-storage capacity through increased buffering. This led to calcium overload in mitochondria, which had two important consequences, both of which resulted in reduced clearance of intracellular calcium after agonist induction: 1) the mitochondria had reached its maximum calcium-storage capacity and hence could not buffer any further calcium released by the ER, and 2) this mitochondrial calcium overload by an unknown mechanism led to inhibition of the sodium–calcium exchanger. This calcium dysregulation is worsened as subsequent calcium influx stimulated by agonist-induced calcium-store depletion (capacitive calcium entry) is augmented in hypoxic astrocytes.Citation35 A similar effect of chronic hypoxic augmentation of agonist-induced calcium release has also been reported in neuroblastoma cells cultured at 2.5% oxygen for 24 hours before experimentation; however, in these cells hypoxia resulted in suppression of the capacitive calcium entry,Citation36 perhaps because calcium signaling was studied in human neurons and recombinant expression systems, as opposed to astrocytes of transgenic rats.
Hypoxia, neuroinflammation, and AD
Neuroinflammation is a key consequence of cerebral hypoxia, and has also been linked to AD pathogenesis. Here, we discuss evidence that cerebral hypoxia can lead to chronic activation and recruitment of proinflammatory immune cells, with particular focus on microglia. This drives production of various mediators/modulators of neuroinflammation, in turn promoting AD pathogenesis. This link between hypoxia, neuroinflammation, and AD has not been adequately reviewed in the literature. This section highlights and organizes critical research evidence for similarities in terms of the substances mediating/modulating the neuroinflammatory environment and the immune cells that drive the formation of these substances.
Inflammation is a localized response by immune cells to tissue injury. Peripheral inflammation has been a well-known process for many centuries, with the first documented description of inflammation as “calor, dolor, rubor, and tumor” by the encyclopedist Celsus in the first century AD. Different cell types are involved in the peripheral inflammatory response: local immune cells (eg, neutrophils) residing at the damaged tissue or immediately entering from the bloodstream give the innate immunoresponse, while other cells (eg, macrophages, T cells, B cells) that reside distant from the damaged tissue (eg, lymph nodes) or show delayed entry from the blood into the damaged tissue give an adaptive immunoresponse. Neuroinflammation is generally characterized by the response of local immune glial cells to damaged tissue, and hence is an innate immunoresponse.
Furthermore, peripheral inflammation can be described as either “acute” or “chronic”. Although the term “acute neuroinflammation” has been presented, this reflects a subtler immunoresponse to limited neuronal insults (such as loss of neuronal efferent or afferents), where the beneficial evolutionarily programmed reparative effects of microglia and astrocytes are fulfilled. Instead, “neuroinflammation” has generally been described as being only a “chronic” process involving sustained cycles of injury and response, whereby chronic immune glial cell (microglial/astrocyte) activation leads to destructive effects. Therefore, here we use the term “neuroinflammation“ to refer to “chronic, CNS-specific, inflammation-like glial responses that do not reproduce the classic characteristics of inflammation in the periphery, but may engender neurodegenerative events”.Citation37
Microglia are the resident macrophages of the CNS, and are the most studied and arguably the most important immune cell mediating neuroinflammation. In a healthy brain, they have a ramified morphology with long, mobile processes that survey the local microenvironment.Citation38 However, upon activation they adopt an amoeboid morphology, allowing them to migrate to the site of the lesion. They are rapidly activated by various pathological triggers, such as protein aggregates (eg, Aβ), cellular debris, pathogenic stimuli, and other factors, such as hypoxia. Recognition of these pathological triggers occur via activation of surface pattern–recognition receptors that bind pathogen-associated molecular patterns and danger-associated molecular patterns. Upon binding, various intracellular signaling cascades are activated, leading to an array of responses involving the production of cytokines/chemokines, acute-phase proteins, and ROS. Evolutionarily, these responses have been programmed to be beneficial by restoring tissue homeostasis; however, in some circumstances, such as chronic activation, the microglial responses are maladaptive (neuroinflammation), resulting in neuronal cell damage and death.Citation39
However, microglial activation is not considered a univalent state: instead, differences in the nature, duration, and strength of the stimulus give rise to different responses.Citation40 Moreover, it has also been shown that there is a diversity of microglia, which gives rise to a variety of responses in the presence of the same stimulus. These different microglial phenotypes have not fully been defined, but may be similar to the key macrophage phenotypes: “proinflammatory” M1 and “anti-inflammatory” M2 cells.Citation41 To determine whether a similar phenotype of microglia is activated in both AD and hypoxic brains, it is important to compare whether the substances produced by these microglia are similar in both cases.
Histological analysis of slices of human AD brains has identified increased levels of morphologically activated microglia. These have been identified in both the white and gray matter, with gray-matter microglia being closely associated with compact plaques containing Aβ.Citation42 In fact, ultrastructural analysis has shown that long processes extending from the microglia surround the Aβ within the compact plaques, suggesting that Aβ itself may play a role in activation of the microglia.Citation42 This observation has also been demonstrated in ultrastructural analysis of transgenic mouse models.Citation42 More recently, it was shown that Toll-like receptors (TLRs; a subtype of pattern-recognition receptors on the surface of the microglia) 2, 4, 6, and 9, alongside CD14 (a coreceptor for TLRs), interact functionally with other microglial surface receptors (such as α6β1 integrin, CD47, CD36, and SCARA1) to bind soluble oligomeric and fibrillar Aβ in plaques and activate intracellular signalling.Citation39,Citation41 It is this intracellular signaling that leads to changes in the morphology of the microglia, and more importantly in the release of various proinflammatory substances that make up the neuroinflammatory environment.
However, it has also been shown that activation of microglia can occur independently of Aβ, and so may be an early component of the disease process. Hayes et al found that microglial load (estimated by analysis of ferritin-immunostained sections of frontal cortex) was unrelated to the age of onset or duration of the disease and did not correlate with Aβ40/42 load.Citation43 Although they found microglial load did correlate with pathological tau levels, the presence of microglial cells precedes the development of pathological tau, and hence the group speculated that the release of proinflammatory molecules from activated microglia may be an initial contributing factor to neurodegeneration. Hayes et al‘s proposal was substantiated by positron-emission tomography imaging, which demonstrated amyloid deposition and microglial activation in patients with mild cognitive impairment (an early symptom of AD) could occur independently of each other.Citation44 These findings suggest that the neuroinflammation resulting from microglial activation may be a similar pathological trigger of AD to Aβ accumulation. Therefore, to identify risk factors underlying sporadic AD, it would be of interest to look at the causes of neuroinflammation, one of which we focus on here — hypoxia.
Activated microglia have been observed in focal and global ischemic animal-brain models and also in human studies.Citation45 Liu et al induced global ischemia for 2.5 or 5 minutes in gerbil brains, and observed a time-dependent increase in levels of 5-bromo-2ʹ-deoxyuridine-5′ (BrdU)-labeled cells (BrdU is able to intercalate into DNA, so is used as a marker for cell proliferation) in the striatum and neocortex. These brdU-labeled cells were mostly identified as activated microglia. Furthermore, Zhang et al showed in focal ischemic rat models that although microglia were absent from the focal ischemic area, there was evidence of amoeboid-shaped cells (activated microglia) in the surrounding areas that contained shrunken neurons.Citation46 These results were also reflected in human studies: Gerhard et al used more advanced technology to measure increased binding of 11C PK1195 (a ligand that binds at the peripheral benzodiazepine-binding site found on activated microglia) in infarcted areas of stroke patients.Citation47
Astrocytes are also important components of the neuroinflammatory response. Postmortem human tissue from AD patients has shown activated, hypertrophic astrocytes close to senile plaques, and this has also been confirmed in animal models of AD.Citation39 This “astrogliosis” is characterized by increased production of GFAP, a protein whose levels also increase after ischemia. In both cases, this astrogliosis is generally described as a proinflammatory maladaptive response.
Blood leukocytes have also been characterized in ischemic brains. After a stroke, increased expression of adhesion molecules on endothelial cells promotes diapedesis of neutrophils, which are then attracted to the ischemic area via chemokines. After neutrophil invasion, monocytes also migrate into ischemic tissue, with peak migration 3–7 days after onset of ischemia. These monocytes then become macrophages and display very similar morphology to activated microglia, thus making them difficult to distinguish. Therefore, the previous experiments describing activated microglia in hypoxic brains may be referring to macrophages. These neutrophils and macrophages have a phagocytic role, but also release substances (similar to astrocytes and microglia) that promote neuroinflammation.Citation48
The role of blood leukocytes in AD-associated neuroinflammation is not known. Animal studies have shown infiltration of blood leukocytes in mice with APs.Citation49 However, in many of these studies, the animals were irradiated to trace blood mononuclear cells and bone-marrow cells. This irradiation may lead to disruption of the blood–brain barrier, which may cause leukocyte infiltration. Further similar experiments where the brain was not irradiated failed to show infiltration of peripheral leukocytes.Citation39 This possible difference in contribution of peripheral leukocytes in neuroinflammation in hypoxic and AD brains can be reconciled by demonstrating similarities in the substances produced by immune cells in both cases.
The neuroinflammatory environment
We have discussed immune cells that drive neuroinflammation being activated in both ischemic and AD brains. To strengthen the link further among AD, hypoxia, and neuroinflammation, it is important to show that the factors released by these immune cells (ie, the mediators and modulators of neuroinflammation) are similar in hypoxic (ischemia-dependent or -independent) and AD brains. These factors are able to promote AD pathogenesis.
Cytokines
Cytokines are a family of proteins produced in largest quantities by immune cells that act on other cells in either a proinflammatory or anti-inflammatory manner. There have been similar reports of several cytokines in both ischemic and AD brains, as hypoxia increases their release from immune cells in an ischemic-dependentCitation50 and -independentCitation51 manner.
In ischemic animal models,IL1β levels show a biphasic release pattern: an immediate peak 1 hour after reperfusion, due to release from activated microglia, and a later peak, due to release from blood-derived monocytes.Citation52 Also, quantitative assays have shown overexpression of IL1β in AD brains, an event that occurs early during plaque development.Citation48 IL1β has been suggested to contribute to the pathogenesis seen in AD in several ways: promotion of amyloidogenic processing and synthesis of APP, activation of microglia and astrocytes, thus increasing production of further proinflammatory substances, and stimulation of calcium influx into neurons, leading to excitotoxic cell death.Citation48
TNFα shows a biphasic release pattern in ischemic mouse models similar to IL1β.Citation52 It is also present in AD brains, as its levels are increased in AD serum, cerebrospinal fluid, cortex, and glial cell cultures after exposure to Aβ.Citation48 The effects of TNFα are both neuroprotective and neurodegenerative. The neurodegenerative aspect involves the ability of TNFα to inhibit glutamate uptake in vitro, activate microglia/astrocytes via surface receptors, and activate intracellular NFκB, thus increasing its own production and production of other proinflammatory substances.Citation48
Low levels of IL6 are present in healthy brains, but many studies have identified an increased presence in AD brains.Citation53 It is also detectable 4 hours after stroke, with levels peaking after a day and remaining detectable for up to 14 days.Citation52 IL6 is a particularly important cytokine in AD pathogenesis in several ways:Citation48 it is a proinflammatory cytokine that leads to neuropathological changes when overexpressed in transgenic mice, causing deficits in avoidance learning, motor impairment, and seizures, and behavioral deficits; it enhances APP synthesis; and a polymorphism in part of the IL6 gene has been linked with delayed development of AD.
Two of the five TGFβ isoforms are increased after stroke: TGFβ1 and TGFβ2.Citation52 These same two isoforms are also detected in AD brains: TGFβ1 is increased in the cerebrospinal fluid and serum of AD cases and has also been detected in plaques, and TGFβ2 levels are also three times higher in AD brains than controls.Citation48 Although TGFβ is well recognized as an anti-inflammatory cytokine, studies have shown that it may promote AD pathogenesis: overexpression of TGFβ1 resulted in accelerated vascular deposition of Aβ in mice expressing human Aβ/APP, and TGFβ1 increased APP expression and ApoE production.Citation48
Chemokines
In cerebral ischemia, the chemokines MCP1, MRF1, and MIP1 are upregulated in the first 3 hours and remain high for at least 6 hours. After reperfusion, other chemokines, such as IL8, are also upregulated.Citation52 These chemokines have been shown to be prominent in neuroinflammatory AD brains. MIP1β was found in activated astrocytes and MCP1 in activated microglia, with MCP1 also being localized to mature senile plaques. Furthermore, microglia from AD autopsies show increases in IL8, MCP1, and MIP1α after addition of Aβ.Citation48 These chemokines may promote AD pathogenesis by promoting immune-cell recruitment, thus amplifying the neuroinflammatory response.Citation48
Free radicals
ROS are increased after hypoxia-induced calcium overload to neurons, and hypoxic-induced neuroinflammation also provides another mechanism for ROS production.Citation52 Hypoxia causes activation of the enzyme iNOS in immune cells, resulting in the formation of nitric oxide (NO). NO can then lead to vasodilation, thus increasing the blood flow in a protective mechanism against ischemia. However, NO can also lead to formation of radicals by reacting with superoxide to give peroxynitirite. In turn, peroxynitirite leads to production of nitrotyrosine-modified proteins, which have been well documented in AD.Citation48
Downstream effects of hypoxia
Previously, we have provided a link that hypoxia, via activation of microglia (and other immune cells) and through the formation of a neuroinflammatory response, could drive AD pathogenesis.Citation39 The next question to ask is: What are the mechanisms mediating hypoxic activation of the immune cells driving neuroinflammation? Here, we highlight some of these mechanisms, with particular focus on activation of various microglial receptors due to the downstream effects of hypoxia. Activation of these receptors resulting in neuroinflammation via microglial activation has also been directly linked to AD pathology.Citation38
ATP
ATP is decreased in isolated hypoxia, due to significant metabolic changes in the HIF1, 5ʹ-AMPK and oxidative phosphorylation pathways.Citation54,Citation55 It is also released into the extracellular space as a result of ischemia-induced tissue injury. Since hypoxia leads to decreased levels of ATP in the cells, this extracellular ATP is unlikely to be due to ATP released from membrane-disrupted cells alone. Instead, another mechanism proposed is that the low levels of ATP released from cells brings about feed-forward ATP-induced ATP release from astrocytes. This ATP is able to bind to the purinergic receptors P2X12 and P2X7, thereby activating microglia and leading to release of proinflammatory molecules.Citation56 ATP-mediated activation of microglia has been linked to AD pathology. Parvathenani et al demonstrated that ATP results in cortical neuronal death by activation of microglial P2X7 receptors in vitro. Furthermore, they showed that P2X7 receptors are upregulated in mouse models of AD on activated microglia and astrocytes surrounding APs.Citation57
HMGB1
HMGB1 is a protein normally localized in the nuclei of cells; however, hypoxic necrotic cell death leads to active release of this protein following 24 hours of anoxia.Citation58 HMGB1 is then able to bind TLR2 and TLR4 on microglial cells, thereby activating them.Citation56 Using novel object-recognition tests, Mazarati et al demonstrated that recombinant HMGB1 resulted in memory deficits in mice, an effect that was abolished in TLR4-knockout mice. The group speculated that this memory deficit, similarly seen in AD patients, was due to HMGB1-induced inflammation via TLR4 activation.Citation59 As such, HMGB1 has been suggested to be a molecular target of preclinical antibody therapy to delay the onset of AD.Citation60
Glutamate
Extracellular glutamate levels are increased in the ischemic brain, due to hypoxia-mediated suppression of glutamate-reuptake transporters found on astrocytes.Citation61 In vitro experiments on rodent hippocampal neurons in tissue culture have also demonstrated that isolated hypoxia (1% oxygen incubator) increases synaptic glutamate transmission and causes neuronal degeneration.Citation62 This glutamate can bind to and activate microglia via AMPA and NMDA receptors in vitro, leading to a proinflammatory response. However, glutamate binding to metabotropic glutamate receptors on microglia in vitro suppresses the microglial inflammatory response.Citation63 Given the preceding, which subtypes of glutamate receptors are expressed in vivo? Although the expression of receptor subtypes has not been fully characterized, studies have shown expression of AMPA and NMDA glutamate receptors in areas of the hippocampus following ischemia.Citation56 Glutamate toxicity has been linked to AD pathology by inducing excitotoxic neuronal cell death via calcium dysregulation.Citation64 Toxicity may also relate to the ability of glutamate to induce neuroinflammation via microglial activation. This is supported by the finding that memantine (NMDA-receptor antagonist) improved memory and attention in Aβ42-injected rats, an effect that may in part be mediated by attenuated microglial activation.Citation65
Zinc
Zinc is normally found at high levels in presynaptic vesicles of certain glutamatergic axon terminals found in the forebrain, hippocampus, and cerebral cortex.Citation66 During ischemia, histochemical studies have shown that levels of vesicular zinc decrease, while microdialysis analysis demonstrated that levels of extracellular zinc increase. This was thought to be mediated by hypoxia, leading to increased calcium in the axonal terminal, resulting in calcium-induced vesicular release.Citation67 Zinc has been shown to activate microglia, a mechanism dependent on upregulation of NADPH oxidase, activation of PARP1, and translocation of NFκB.Citation56 Zinc has been linked directly to AD pathology, as zinc buffering has given encouraging results in mediating neuroprotection in human stroke victims.Citation68 Although this protection may relate to other neurotoxic mechanisms of zinc, such as direct neuronal excitotoxicity and Aβ precipitation, it may also relate to the ability of zinc to induce neuroinflammation via microglial activation.Citation69
Intermittent hypoxia
We have presented hypoxia as uniformly detrimental to the brain, yet there is increasing evidence that moderate and/or intermittent hypoxia evokes protective adaptations in the CNS. In many of the cited in vitro studies, cell lines were exposed to chronic hypoxia, typically <5% oxygen for >24 hours. In contrast, in ischemic models of stroke, the hypoxia was more acute, lasting minutes to hours. This suggests that for isolated hypoxia, a chronic pattern provokes AD, whereas the more severe ischemia-dependent hypoxia can be more short-lived before causing neuroinflammation and dementia. This is supported by sparse studies of populations living at altitude and hence exposed to chronic hypoxia: analysis in Californian counties suggests that altitude of residence was correlated with the risk of dying of ADCitation70 and neuroinflammation-dependent cognitive changes have been reported during prolonged stays at high altitude.Citation71 However, ecological studies like these must be interpreted cautiously, as it is difficult reliably to control many confounding variables, such as comorbidities and air pollution.
As such, if chronic hypoxia is linked to AD, can the same be said of isolated acute hypoxia? In fact, there is growing evidence that intermittent hypoxia evokes neuroprotective adaptations in the CNS.Citation72 This neuroprotection has manifested as improved cognitive function in elderly people breathing a hypoxic air mixture for 40 minutes three times a weekCitation73 and as reduced stroke-lesion volume in rats exposed to 7 days of 12% oxygen for 4 hours per day.Citation74 Furthermore, rats exposed to intermittent hypoxia for 4 hours daily for 14 days had reduced nitrites and nitrates in the plasma induced by Aβ in experimental AD models.Citation75
However, patients with obstructive sleep apnea stop breathing repeatedly for at least 10 seconds during their sleep, also resulting in intermittent hypoxia. There is evidence of a wide range of cognitive deficits identified among untreated obstructive sleep–apnea patients, from sustained attention to working memory.Citation76 As such, there is a strong association between intermittent hypoxia‐related pathological mechanisms and onset of memory and cognitive dysfunction, which may progress to AD.Citation77
These contradictory findings suggest further research is needed in this area. Discrepancies may be due to a lack of a standardized definition for intermittent hypoxia:Citation72 studies have differences in the severity of hypoxic stimuli and the frequency of hypoxic episodes per day, blurring the lines between therapeutic effects and pathogenesis. Differences also arise from studies using both isolated hypoxia (exposure to low partial pressure of oxygen) and ischemia-dependent hypoxia (occluding or severing arteries). If researchers optimize the balance between efficacy and safety for intermittent hypoxia use in the CNS, it may represent a simple, safe, nonpharmacological method to protect against cognitive decline.
Conclusion
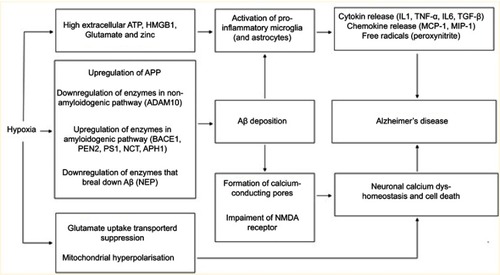
We have provided a body of evidence to highlight key links between hypoxia and AD. Due to altered proteolytic cleavage of APP, Aβ accumulation is the initial pathological trigger of AD, as per the ACH. A key mechanism by which Aβ leads to the pathology seen in AD is by dysregulation of calcium homeostasis (in neurons and astrocytes), resulting in neuronal cell death. Aβ can also trigger activation of microglia, leading to a maladaptive neuroinflammatory response that further contributes to AD pathology. However, evidence has shown that the neuroinflammatory response in AD brains can also be triggered independently of Aβ. Therefore, neuroinflammation itself could be considered another initiating pathological trigger of AD. Hypoxia is not only able to promote Aβ formation and accumulation but independently of Aβ can also dysregulate calcium homeostasis (in both neurons and astrocytes), leading to neuronal cell death and activate microglia and result in a neuroinflammatory response ().
Figure 1 A summary of the various links between hypoxia and AD (both dependent and independent of Aβ accumulation) discussed..
Abbreviations: ATP, Adenosine triphosphate; HMGB1, High mobility group box 1; APP, Amyloid precursor protein; ADAM10, A disintegrin and metalloproteinase domain-containing protein 10; BACE1, Beta-secretase 1; PEN2, Presenilin enhancer 2; PS1, Presenilin 1; NCT, Nicastrin; APH1, Anterior pharynx-defective 1; NEP, Neprilysin; AB, Amyloid-β; NMDA, N-methyl-D-aspartate; IL1, Interleukin-1; TNF-α, Tumor necrosis factor α; IL6, Interleukin-6; TGF-B, Transforming growth factor-β; MCP-1, Monocyte chemoattractant protein-1; MIP-1, Macrophage inflammatory protein-1.
Therapy for AD is very limited, with acetylcholine esterase inhibitors (donepezil, galantamine) and NMDA-receptor antagonists (memantine) being the only medications available in the UK. These medications are beneficial in reducing the symptoms of the disease; however, they have little effect on improving disease pathology. Understanding the links between acute/chronic hypoxia and AD is important, as until more advanced therapy becomes available, preventive measures to prevent chronic cerebral hypoxia may not only help prevent the development of AD but may also benefit patients who already have this disease.
Disclosure
The authors report no conflicts of interest in this work.
References
- Prince M, Knapp M, Guerchet M, et al. Dementia UK: Update. 2nd ed U.K: Alzheimer’s Society; 2014 Available from: https://www.alzheimers.org.uk/info/20025/policy_and_influencing/251/dementia_uk.
- Lancet T. Dementia burden coming into focus. Lancet. 2017;390(10113):2606. doi:10.1016/S0140-6736(17)33304-4
- Dementia. Available from: https://www.who.int/news-room/fact-sheets/detail/dementia. Accessed 224, 2019.
- Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–185.1566067
- LaFerla FM Amyloid-β and tau in Alzheimer’s disease. [updated 5, 2008] Available from: http://www.nature.com/nrn/posters/ad/index.html. Accessed 415, 2017.
- Mudher A, Lovestone S. Alzheimer’s disease-do tauists and baptists finally shake hands? Trends Neurosci. 2002;25(1):22–26.11801334
- Shi J, Yang SH, Stubley L, Day AL, Simpkins JW. Hypoperfusion induces overexpression of beta-amyloid precursor protein mRNA in a focal ischemic rodent model. Brain Res. 2000;853(1):1–4.10627301
- Hall ED, Oostveen JA, Dunn E, Carter DB. Increased amyloid protein precursor and apolipoprotein E immunoreactivity in the selectively vulnerable hippocampus following transient forebrain ischemia in gerbils. Exp Neurol. 1995;135(1):17–27. doi:10.1006/exnr.1995.10627556550
- Mattson MP. Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev. 1997;77(4):1081–1132.9354812
- Webster NJ, Green KN, Settle VJ, Peers C, Vaughan PFT. Altered processing of the amyloid precursor protein and decreased expression of ADAM 10 by chronic hypoxia in SH-SY5Y: no role for the stress-activated JNK and p38 signalling pathways. Brain Res Mol Brain Res. 2004;130(1–2):161–169. doi:10.1016/j.molbrainres.2004.06.04215519686
- Auerbach ID, Vinters HV. Effects of anoxia and hypoxia on amyloid precursor protein processing in cerebral microvascular smooth muscle cells. J Neuropathol Exp Neurol. 2006;65(6):610–620.16783171
- Rybnikova E, Gluschenko T, Galeeva A, et al. Differential expression of ADAM15 and ADAM17 metalloproteases in the rat brain after severe hypobaric hypoxia and hypoxic preconditioning. Neurosci Res. 2012;72(4):364–373. doi:10.1016/j.neures.2011.12.01022230263
- Sun X, He G, Qing H, et al. Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc Natl Acad Sci U S A. 2006;103(49):18727–18732. doi:10.1073/pnas.060629810317121991
- Peers C, Dallas ML, Boycott HE, Scragg JL, Pearson HA, Boyle JP. Hypoxia and neurodegeneration. Ann N Y Acad Sci. 2009;1177:169–177. doi:10.1111/j.1749-6632.2009.05026.x19845619
- Wang R, Zhang Y-W, Zhang X, et al. Transcriptional regulation of APH-1A and increased gamma-secretase cleavage of APP and Notch by HIF-1 and hypoxia. FASEB J Off Publ Fed Am Soc Exp Biol. 2006;20(8):1275–1277. doi:10.1096/fj.06-5839fje
- Pluta R, Jabłoński M, Ułamek-Kozioł M, et al. Sporadic Alzheimer’s disease begins as episodes of brain ischemia and ischemically dysregulated Alzheimer’s disease genes. Mol Neurobiol. 2013;48(3):500–515. doi:10.1007/s12035-013-8439-123519520
- Liu H, Qiu H, Yang J, Ni J, Le W. Chronic hypoxia facilitates Alzheimer’s disease through demethylation of γ-secretase by downregulating DNA methyltransferase 3b. Alzheimers Dement J Alzheimers Assoc. 2016;12(2):130–143. doi:10.1016/j.jalz.2015.05.019
- Kerridge C, Kozlova DI, Nalivaeva NN, Turner AJ. Hypoxia affects neprilysin expression through caspase activation and an APP intracellular domain-dependent mechanism. Front Neurosci. 2015;9:426. doi:10.3389/fnins.2015.0042626617481
- Wang Z, Zhang X-J, Li T, Li J, Tang Y, Le W. Valproic acid reduces neuritic plaque formation and improves learning deficits in APP(Swe)/PS1(A246E) transgenic mice via preventing the prenatal hypoxia-induced down-regulation of neprilysin. CNS Neurosci Ther. 2014;20(3):209–217. doi:10.1111/cns.1218624289518
- Perl DP. Neuropathology of Alzheimer’s disease. Mt Sinai J Med N Y. 2010;77(1):32–42. doi:10.1002/msj.20157
- Supnet C, Bezprozvanny I. Neuronal calcium signaling, mitochondrial dysfunction, and Alzheimer’s disease. J Alzheimers Dis JAD. 2010;20(Suppl 2):S487–S498. doi:10.3233/JAD-2010-10030620413848
- Arispe N, Rojas E, Pollard HB. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc Natl Acad Sci U S A. 1993;90(2):567–571.8380642
- Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430(7000):631–639. doi:10.1038/nature0262115295589
- Cha M-Y, Han S-H, Son SM, et al. Mitochondria-specific accumulation of amyloid β induces mitochondrial dysfunction leading to apoptotic cell death. PLoS One. 2012;7(4):e34929. doi:10.1371/journal.pone.003492922514691
- Kadowaki H, Nishitoh H, Urano F, et al. Amyloid beta induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 2005;12(1):19–24. doi:10.1038/sj.cdd.440152815592360
- Troy CM, Rabacchi SA, Friedman WJ, Frappier TF, Brown K, Shelanski ML. Caspase-2 mediates neuronal cell death induced by beta-amyloid. J Neurosci Off J Soc Neurosci. 2000;20(4):1386–1392. doi:10.1523/JNEUROSCI.20-04-01386.2000
- Abramov AY, Canevari L, Duchen MR. Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J Neurosci Off J Soc Neurosci. 2003;23(12):5088–5095. doi:10.1523/JNEUROSCI.23-12-05088.2003
- Kimelberg HK, Nedergaard M. Functions of astrocytes and their potential as therapeutic targets. Neurotherapeutics. 2010;7(4):338–353. doi:10.1016/j.nurt.2010.07.00620880499
- Green KN, Peers C. Amyloid beta peptides mediate hypoxic augmentation of Ca(2+) channels. J Neurochem. 2001;77(3):953–956.11331424
- Webster NJ, Ramsden M, Boyle JP, Pearson HA, Peers C. Amyloid peptides mediate hypoxic increase of L-type Ca2+ channels in central neurones. Neurobiol Aging. 2006;27(3):439–445. doi:10.1016/j.neurobiolaging.2005.02.00216464656
- Scragg JL, Fearon IM, Boyle JP, Ball SG, Varadi G, Peers C. Alzheimer’s amyloid peptides mediate hypoxic up-regulation of L-type Ca2+ channels. FASEB J. 2005;19(1):150–152. doi:10.1096/fj.04-2659fje15494446
- Boycott HE, Dallas M, Boyle JP, Pearson HA, Peers C. Hypoxia suppresses astrocyte glutamate transport independently of amyloid formation. Biochem Biophys Res Commun. 2007;364(1):100–104. doi:10.1016/j.bbrc.2007.09.10217927959
- Kristián T, Siesjö BK. Calcium in ischemic cell death. Stroke. 1998;29(3):705–718.9506616
- Smith IF, Boyle JP, Plant LD, Pearson HA, Peers C. Hypoxic remodeling of Ca2+ stores in type i cortical astrocytes. J Biol Chem. 2003;278(7):4875–4881. doi:10.1074/jbc.M20920620012477727
- Smith IF, Plant LD, Boyle JP, Skinner RA, Pearson HA, Peers C. Chronic hypoxia potentiates capacitative Ca2+ entry in type-I cortical astrocytes. J Neurochem. 2003;85(5):1109–1116.12753070
- Smith IF, Boyle JP, Vaughan PF, Pearson HA, Peers C. Effects of chronic hypoxia on Ca(2+) stores and capacitative Ca(2+) entry in human neuroblastoma (SH-SY5Y) cells. J Neurochem. 2001;79(4):877–884.11723180
- Streit WJ, Mrak RE, Griffin WST. Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation. 2004;1:14. doi:10.1186/1742-2094-1-1415285801
- Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med. 2017;23(9):1018–1027. doi:10.1038/nm.439728886007
- Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388–405. doi:10.1016/S1474-4422(15)70016-525792098
- Xing C, Arai K, Lo EH, Hommel M. Pathophysiologic cascades in ischemic stroke. Int J Stroke Off J Int Stroke Soc. 2012;7(5):378–385. doi:10.1111/j.1747-4949.2012.00839.x
- Cameron B, Landreth GE. Inflammation, microglia, and Alzheimer’s disease. Neurobiol Dis. 2010;37(3):503–509. doi:10.1016/j.nbd.2009.10.00619833208
- Combs CK. Inflammation and microglia actions in Alzheimer’s disease. J Neuroimmune Pharmacol off J Soc Neuroimmune Pharmacol. 2009;4(4):380–388. doi:10.1007/s11481-009-9165-3
- Hayes A, Thaker U, Iwatsubo T, Pickering-Brown SM, Mann DMA. Pathological relationships between microglial cell activity and tau and amyloid beta protein in patients with Alzheimer’s disease. Neurosci Lett. 2002;331(3):171–174.12383924
- Okello A, Edison P, Archer HA, et al. Microglial activation and amyloid deposition in mild cognitive impairment: a PET study. Neurology. 2009;72(1):56–62. doi:10.1212/01.wnl.0000338622.27876.0d19122031
- Liu J, Bartels M, Lu A, Sharp FR. Microglia/macrophages proliferate in striatum and neocortex but not in hippocampus after brief global ischemia that produces ischemic tolerance in gerbil brain. J Cereb Blood Flow Metab. 2001;21(4):361–373. doi:10.1097/00004647-200104000-0000511323522
- Zhang Z, Chopp M, Powers C. Temporal profile of microglial response following transient (2 h) middle cerebral artery occlusion. Brain Res. 1997;744(2):189–198. doi:10.1016/S0006-8993(96)01085-29027378
- Gerhard A, Neumaier B, Elitok E, et al. In vivo imaging of activated microglia using [11C]PK11195 and positron emission tomography in patients after ischemic stroke. Neuroreport. 2000;11(13):2957–2960.11006973
- Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21(3):383–421.10858586
- Hohsfield LA, Humpel C. Migration of blood cells to β-amyloid plaques in Alzheimer’s disease. Exp Gerontol. 2015;65:8–15. doi:10.1016/j.exger.2015.03.00225752742
- Bona E, Andersson AL, Blomgren K, et al. Chemokine and inflammatory cell response to hypoxia-ischemia in immature rats. Pediatr Res. 1999;45(4 Pt 1):500–509. doi:10.1203/00006450-199904010-0000810203141
- Naldini A, Carraro F, Silvestri S, Bocci V. Hypoxia affects cytokine production and proliferative responses by human peripheral mononuclear cells. J Cell Physiol. 1997;173(3):335–342. doi:10.1002/(SICI)1097-4652(199712)173:3<335::AID-JCP5>3.0.CO;2-O9369946
- Ceulemans A-G, Zgavc T, Kooijman R, Hachimi-Idrissi S, Sarre S, Michotte Y. The dual role of the neuroinflammatory response after ischemic stroke: modulatory effects of hypothermia. J Neuroinflammation. 2010;7:74. doi:10.1186/1742-2094-7-7421040547
- Rothaug M, Becker-Pauly C, Rose-John S. The role of interleukin-6 signaling in nervous tissue. Biochim Biophys Acta BBA - Mol Cell Res. 2016;1863(6, Part A):1218–1227. doi:10.1016/j.bbamcr.2016.03.018
- Solaini G, Baracca A, Lenaz G, Sgarbi G. Hypoxia and mitochondrial oxidative metabolism. Biochim Biophys Acta. 2010;1797(6–7):1171–1177. doi:10.1016/j.bbabio.2010.02.01120153717
- Wheaton WW, Chandel NS. Hypoxia. 2. Hypoxia regulates cellular metabolism. Am J Physiol - Cell Physiol. 2011;300(3):C385–C393. doi:10.1152/ajpcell.00485.201021123733
- Yenari MA, Kauppinen TM, Swanson RA. Microglial activation in stroke: therapeutic targets. Neurother J Am Soc Exp Neurother. 2010;7(4):378–391. doi:10.1016/j.nurt.2010.07.005
- Parvathenani LK, Tertyshnikova S, Greco CR, Roberts SB, Robertson B, Posmantur R. P2X7 mediates superoxide production in primary microglia and is up-regulated in a transgenic mouse model of Alzheimer’s disease. J Biol Chem. 2003;278(15):13309–13317. doi:10.1074/jbc.M20947820012551918
- Faraco G, Fossati S, Bianchi ME, et al. High mobility group box 1 protein is released by neural cells upon different stresses and worsens ischemic neurodegeneration in vitro and in vivo. J Neurochem. 2007;103(2):590–603. doi:10.1111/j.1471-4159.2007.04788.x17666052
- Mazarati A, Maroso M, Iori V, Vezzani A, Carli M. High-mobility group box-1 impairs memory in mice through both toll-like receptor 4 and receptor for advanced glycation end products. Exp Neurol. 2011;232(2):143–148. doi:10.1016/j.expneurol.2011.08.01221884699
- Fujita K, Motoki K, Tagawa K, et al. HMGB1, a pathogenic molecule that induces neurite degeneration via TLR4-MARCKS, is a potential therapeutic target for Alzheimer’s disease. Sci Rep. 2016;6:31895. doi:10.1038/srep3189527557632
- Kobayashi S, Millhorn DE. Hypoxia regulates glutamate metabolism and membrane transport in rat PC12 cells. J Neurochem. 2001;76(6):1935–1948.11259512
- Choi DW, Rothman SM. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu Rev Neurosci. 1990;13:171–182. doi:10.1146/annurev.ne.13.030190.0011311970230
- Spampinato SF, Copani A, Nicoletti F, Sortino MA, Caraci F. Metabotropic glutamate receptors in glial cells: a new potential target for neuroprotection? Front Mol Neurosci. 2018;11. doi:10.3389/fnmol.2018.00414.
- Danysz W, Parsons CG. Alzheimer’s disease, β-amyloid, glutamate, NMDA receptors and memantine – searching for the connections. Br J Pharmacol. 2012;167(2):324–352. doi:10.1111/j.1476-5381.2012.02057.x22646481
- Nyakas C, Granic I, Halmy LG, Banerjee P, Luiten PGM. The basal forebrain cholinergic system in aging and dementia. Rescuing cholinergic neurons from neurotoxic amyloid-β42 with memantine. Behav Brain Res. 2011;221(2):594–603. doi:10.1016/j.bbr.2010.05.03320553766
- Sandstead HH, Frederickson CJ, Penland JG. History of zinc as related to brain function. J Nutr. 2000;130(2):496S–502S. doi:10.1093/jn/130.2.496S10721938
- Kim S, Seo J-W, Oh SB, et al. Disparate roles of zinc in chemical hypoxia-induced neuronal death. Front Cell Neurosci. 2015;9. doi:10.3389/fncel.2015.00001
- Frederickson CJ, Cuajungco MP, Frederickson CJ. Is zinc the link between compromises of brain perfusion (excitotoxicity) and Alzheimer’s disease? J Alzheimers Dis JAD. 2005;8(2):155–160; discussion 209–215.
- Kauppinen TM, Higashi Y, Suh SW, Escartin C, Nagasawa K, Swanson RA. Zinc triggers microglial activation. J Neurosci Off J Soc Neurosci. 2008;28(22):5827–5835. doi:10.1523/JNEUROSCI.1236-08.2008
- Thielke S, Slatore CG, Banks WA. Association between Alzheimer dementia mortality rate and altitude in California counties. JAMA Psychiatry. 2015;72(12):1253–1254. doi:10.1001/jamapsychiatry.2015.185226502029
- Hu SL, Xiong W, Dai ZQ, Zhao HL, Feng H. Cognitive changes during prolonged stay at high altitude and its correlation with C-reactive protein. PLoS One. 2016;11(1). doi:10.1371/journal.pone.0146290
- Navarrete-Opazo A, Mitchell GS. Therapeutic potential of intermittent hypoxia: a matter of dose. Am J Physiol - Regul Integr Comp Physiol. 2014;307(10):R1181–R1197. doi:10.1152/ajpregu.00208.201425231353
- Schega L, Peter B, Törpel A, Mutschler H, Isermann B, Hamacher D. Effects of intermittent hypoxia on cognitive performance and quality of life in elderly adults: a pilot study. Gerontology. 2013;59(4):316–323. doi:10.1159/00035092723652274
- Tsai YW, Yang YR, Chen GH, Chang HC, Wang RY. The time window of intermittent hypoxia intervention after middle cerebral artery occlusion. Chin J Physiol. 2008;51(5):324–328.19175189
- Mashina SY, Aleksandrin VV, Goryacheva AV, et al. Adaptation to hypoxia prevents disturbances in cerebral blood flow during neurodegenerative process. Bull Exp Biol Med. 2006;142(2):169–172. doi:10.1007/s10517-006-0318-617369930
- Ferini-Strambi L, Lombardi GE, Marelli S, Galbiati A. Neurological deficits in obstructive sleep apnea. Curr Treat Options Neurol. 2017;19(4):16. doi:10.1007/s11940-017-0451-828374233
- Daulatzai MA. Evidence of neurodegeneration in obstructive sleep apnea: relationship between obstructive sleep apnea and cognitive dysfunction in the elderly. J Neurosci Res. 2015;93(12):1778–1794. doi:10.1002/jnr.2363426301370