Abstract
Perampanel (PER) is a novel antiepileptic compound that decreases neuronal excitability by modulating glutamatergic transmission through selective noncompetitive blockade of AMPA receptors. PER has been evaluated in three pivotal placebo-controlled randomized trials as adjunctive therapy in adult drug-resistant partial epilepsy. In comparison to placebo, adjunctive PER effectively reduces seizure frequency. The relative risk of the responder rate (95% confidence interval [CI]) was thus 1.60 (1.08–2.36), 1.79 (1.42–2.25) and 1.66 (1.24–2.23) for once-daily PER 4 mg/day, 8 mg/day and 12 mg/day, respectively. The most common adverse events associated with PER were nonspecific central nervous system side effects. Some concerns have been raised about risk of clinically significant weight gain and of psychiatric adverse events. Long-term open-label extensions of the three pivotal trials are underway. PER has recently been approved both in Europe and in the USA for the adjunctive treatment of partial onset seizures in patients aged 12 years and above. However, in the absence of a direct comparison between PER and other licensed antiepileptic drugs’ efficacy and tolerability, the clinical advantages of PER over the other drugs in intractable partial epilepsy remains to be determined.
Introduction
Over the past 20 years, the armamentarium of antiepileptic drugs (AEDs) has exponentially increased. In addition to the five standard AEDs developed before 1970, phenobarbital, phenytoin, carbamazepine, sodium valproate, ethosuximide, and 14 other compounds are now available for the treatment of adult partial epilepsy. However, their impact on the proportion of patients whose partial seizures remain uncontrolled has been limited.Citation1 While 64% of patients with newly-diagnosed epilepsy achieved seizure-free status in the Glasgow’s cohort in 2000,Citation2 this proportion only increased to 68% ten years later.Citation3 On the other hand, new AEDs are associated with a wider adverse effects profile and frequently show improved tolerability in comparison to standard AEDs.Citation4 The increasing number of AEDs has thus provided better individualization of the treatment to the patients’ characteristics, especially with regard to side effects, patients’ sex, comorbidities and drug interactions.Citation1
Although the mechanisms of epileptogenesis and AED resistance appear highly heterogeneous,Citation5,Citation6 the variety of mechanisms of action of licensed AEDs remains limited. Indeed, most AEDs exert their primary antiepileptic action through one of the following molecular pathways:Citation7 (1) inhibition of sustained repetitive neuronal firing via the blockade or modulation of voltage-gated sodium or potassium channels, respectively, (2) increased GABAergic transmission by inhibiting GABA recapture or metabolism, or allosteric modulation of postsynaptic GABAA receptors, and (3) decreased vesicular release through modulation of presynaptic calcium channels or synaptic vesicle 2A protein. Although the multifactorial nature of drug resistance in epilepsy makes it improbable that a single drug could eradicate refractory epilepsy, the development of AEDs with novel molecular targets and mechanisms of action offers opportunity to improve both seizure control and treatment tolerability. Indeed, polytherapy with drugs sharing similar mechanisms of action carries the risk of increased neurotoxicity.Citation8
AMPA receptors and seizure generation
Glutamate is the main excitatory neurotransmitter in the central nervous system. It acts on both ionotropic and metabotropic receptors. Ionotropic glutamate receptors underlie fast synaptic excitatory transmission and are classified into three major types: AMPA, NMDA and kainate.Citation9 AMPA receptors are the most abundant. Binding of glutamate opens AMPA receptors, which allows cations, mainly sodium, to enter into the cell.Citation9 This cation influx results in a brief depolarization of the postsynaptic membrane. Summation of excitatory postsynaptic potentials leads to the firing of action potentials by the postsynaptic neuron. NMDA receptors are permeable to calcium as well as sodium and potassium.Citation9 However, they are blocked by magnesium at resting membrane potential and their activation requires sufficient depolarization, mostly provided by activation of AMPA receptors, to relieve magnesium block.Citation9 NMDA-related calcium influx triggers several forms of synaptic plasticity, including long-term potentiation which strengthens synaptic signaling.
In animal models of epilepsy, activation of both NMDA and AMPA receptors play an important role in seizure generation, making it difficult to isolate the action of one receptor subtype. In most slice models, it is necessary to block both NMDA and AMPA receptors to fully suppress epileptiform activities.Citation10–Citation12 However, in other models, antagonists of AMPA receptors alone nearly suppressed all discharges.Citation13 Furthermore, NMDA antagonists might paradoxically enhance the frequency of bursting at the same time that they reduce the burst duration and the number of spikes in each discharge.Citation14 Finally, epileptic discharges observed in human neocortical tissue removed during epilepsy surgery were found to be more sensitive to AMPA than to NMDA receptor blockers.Citation15
In this context, developing drugs that would specifically target AMPA receptors might be of particular interest in the treatment of epilepsy, especially since the development of selective NMDA antagonists failed because of prominent side effects and/or poor efficacy.Citation16 Two selective AMPA antagonists, talampanel and perampanel, have been evaluated in humans. Although talampanel has been positively evaluated in a Phase II trial as an add-on therapy in drug-resistant partial epilepsy,Citation17 its relatively short half-life has limited its clinical development.Citation18 In contrast, perampanel (PER) has recently been approved in the US and Europe as an add-on treatment for drug-resistant partial epilepsy.
Pharmacological profile
Effect of perampanel on AMPA receptors
PER is a noncompetitive, selective antagonist of the AMPA receptor. In rat cortical neurons, PER inhibited AMPA-induced increases in [Ca2+]i in a concentration-dependent manner.Citation19 Similarly, PER inhibited AMPA receptor-mediated field excitatory postsynaptic potentials in hippocampal slices.Citation20 In contrast, PER had no significant effect, either on responses mediated by NMDA receptors or on those mediated by kainate receptors.Citation19,Citation20 Radiolabeled PER, [3H] perampanel, was used to assess binding specificity, and demonstrated a noncompetitive interaction between PER and AMPA receptors. Thus, [3H] PER binding to rat forebrain membranes was not significantly reduced by glutamate (1 mM), AMPA (0.1 mM), or by the competitive glutamate receptor antagonist NBQX (0.1 mM).Citation19 In contrast, [3H] PER binding was displaced by the noncompetitive AMPA receptor antagonists CP465022 and GYKI52466.Citation19 These results suggested that PER might share the same binding site as these two compounds, the latter being at the interface between the glutamate binding core and the channel region of the AMPA receptor.Citation21 This binding site might stabilize the resting state of the channel by disrupting channel opening in response to agonist binding.Citation21
Effect of perampanel in animal models of epilepsy
PER has exhibited antiseizure effects in several animal models of acute seizures. PER prevented tonic-clonic generalized seizures in both audiogenic and maximal electroshock seizure tests, and absence or myoclonic seizures in PTZ-induced seizure tests.Citation19 Similarly, PER protected mice in a dose-dependent manner from 6 Hz electroshock-induced seizures, and significantly increased the threshold of after discharges in amygdala-kindled rats.Citation19 However, PER efficacy on spontaneous seizures in animal models of chronic epilepsy has not been reported yet.
Clinical pharmacokinetics and interactions
Following oral administration, PER is rapidly and almost completely absorbed, with peak plasma concentrations reached at 15 minutes to 2 hours after dosing ().Citation22 Bioavailability approaches 100%,Citation22 and the protein binding is 95%. The elimination half-life is long, estimated around 70 hours, allowing a once-daily regimen.Citation22 PER is primarily eliminated by hepatic oxidative metabolism. About 70% of the dose is excreted in the feces whereas less than 2% is excreted unchanged in the urine. PER metabolism is primarily supported by cytochrome P450 (CYP)-3A4.Citation23 However, the involvement of other enzymes has not been ruled out, since the metabolic pathway responsible for the production of some metabolites remains unknown. The metabolism of PER is associated with the formation of reactive intermediates whose impact also remains unknown.
Table 1 Pharmacokinetics of perampanel
According to its main metabolic pathway, PER pharmacokinetics are influenced by enzyme-inducing drugs, and specifically by CYP3A4 inducers.Citation23 As a matter of fact, treatment with carbamazepine increased the clearance of PER 3-fold whereas the CYP3A4 inhibitor ketoconazole increased its bioavailability by 20%. In vitro studies showed that PER had a small inhibitory effect on CYP2C8 and no effect on CYP3A4. Doses of 4 mg and 8 mg of PER did not alter the pharmacokinetics of oral contraception components, ethinyl estradiol and levonorgestrel.Citation23 However, it has been shown that the administration of 12 mg PER reduced levonorgestrel bioavailability by approximately 40%, while having no effect on ethinyl estradiol bioavailability.
A prospective study evaluated the modifications of PER pharmacokinetics in subjects with hepatic impairment. Elimination half-life was increased by about 2-fold in subjects with mild to moderate hepatic dysfunction.Citation23 No study has been conducted in patients with severe hepatic impairment or with impaired renal function. Importantly, there was no effect of age on the pharmacokinetics of PER so that no dose adjustment is needed for the elderly.
Clinical efficacy
Because it would be unethical to treat patients with epilepsy in monotherapy with a drug whose clinical efficacy is unknown, new AEDs are initially evaluated as adjunctive therapy in patients with drug-resistant epilepsy.Citation24 In addition, localization-related epilepsies being the most prevalent refractory seizure disorder in adults, these trials are usually conducted in patients with drug-resistant partial seizures.Citation24 The patients included in these studies have uncontrolled partial onset seizures despite treatment with a minimum of two to three different AEDs. To date, PER has only been evaluated as add-on therapy in patients with refractory partial epilepsy in two Phase-II randomized controlled trials (RCTs)Citation25 and in three pivotal Phase-III placebo-controlled RCTs.Citation26–Citation28 In addition, interim results of the long-term open-label extension of the two Phase-II studies as well as interim results of the open-label extension phase of the Phase-III RCTs were recently published.Citation29,Citation30 A study evaluating PER as adjunctive therapy in refractory primary generalized tonic-clonic seizures is ongoing. No monotherapy study has been launched so far.
As in all previous adjunctive-therapy RCTs of second generation AEDs, the primary efficacy endpoints were the reduction from baseline in seizure frequency per 28 days and/or the responder rate, which corresponded to the proportion of patients who achieved 50% or greater reduction in seizure frequency as compared to baseline. Responder rates were primarily calculated on an intent-to-treat basis using the last observation carried forward analysis, in which patients who withdrew prematurely, including withdrawals due to adverse effects, that experienced greater than 50% reduction in seizures during the time they were in the treatment period, are counted as responders. Importantly, it has been demonstrated that last observation carried forward analysis overestimates, in a dose-dependent manner, the responder rates in comparison with the method whereby only study completers are counted as responders.Citation31 Accordingly it has been recommended that the reports of Phase-III trials should present both last observation carried forward and completer analyses to improve their clinical relevance.Citation31,Citation32 However, the reports of PER RCTs did not provide this information and the efficacy data presented in the current review were obtained using the last observation carried forward method. The only exception was the seizure free rate that was calculated with the method whereby only the study completers who were seizure-free during the entire maintenance period were counted as seizure-free patients.Citation32
Phase-II studies
Two Phase-II randomized dose-escalation studies have been published.Citation25 Both studies included adult patients aged 18–70 years. All patients were Caucasian in study 208, whereas 13% were non-Caucasian in study 206. In study 206, patients received placebo or PER up to 4 mg/day either dosed once-daily or twice-daily. Titration period lasted 8 weeks and was followed by a 4-week maintenance phase. In study 208, once-daily PER could be titrated up to 12 mg/day over a period of 8 weeks before patients entered a 4-week maintenance phase. However, in both studies, if a patient did not tolerate treatment during titration, the dose could be reduced to the previous level. In study 206, 82.4% of patients reached and tolerated PER 4 mg/day. In study 208, it was estimated that 97% of patients tolerated PER 4 mg/day, 55% PER 8 mg/day and 44% PER 12 mg/day. Although the primary endpoint of both studies was PER tolerability, the 50% responder rate was also provided. However, according to limited sample size, efficacy endpoints should be interpreted with caution. Study 206 included 153 patients, 51 randomized to placebo, and 102 to PER (51 to once-daily PER and 51 to twice-daily PER). The responder rate was 30.7% with PER and 21.6% with placebo (P = 0.19). Study 208 included 48 patients, ten randomized to placebo and 38 to PER. As in study 206, the responder rate did not significantly differ between placebo (22.2%) and PER groups (39.5%).
Phase-III studies
Three Phase-III studies have been published ().Citation26–Citation28 All were multicenter, randomized, double-blind, parallel-group trials evaluating the efficacy and safety of PER in comparison to placebo in the adjunctive treatment of refractory partial epilepsy.
Table 2 Clinical efficacy in Phase III RCTs
Studies 304 and 305 had identical trial designs. They only differed by the location of the study centers. Study 304 was conducted in North America (US and Canada), Central America (Mexico) and South America (Argentina and Chile)Citation27 whereas Study 305 was conducted in the European Union, in the US, Israel, Russia, UK, India, Australia and South Africa.Citation26 Following a prospective 6-week baseline phase, patients entered a 19-week double-blind treatment period that was divided into a 6-week titration phase and a 13-week maintenance phase. Both studies assessed the efficacy and safety of once-daily PER 8 mg/day and 12 mg/day.Citation26,Citation27 However, patients experiencing intolerable adverse events could stay on the same dose or have their dose reduced. Baseline patients’ characteristics were similar between the two studies and well balanced across study groups. Patients were on average 36 years of age (range 12–77). The mean duration of epilepsy was approximately 23 years and the median baseline partial onset seizure frequency per month was 13.7 and 11.8 in the placebo arms and 13.2 and 13.4 in the PER arms. Patients received one to three other AEDs.
A total of 388 patients were randomized in study 304, and 389 in study 305. 133 and 129 were assigned to PER 8 mg/day in studies 304 and 305, respectively, and 134 and 121 to PER 121 mg/day. 121 patients received placeo in study 304 and 136 in study 305. The proportions of patients achieving their randomized dose in study 304 were 103 (77.4%) and 78 (58.7%) for 8 and 12 mg PER, respectively. In study 305, 99 patients achieved 8 mg PER, nine achieved 10 mg PER and 70 achieved 12 mg PER. Median percentages of seizure reduction were greater in PER-treated patients than in placebo-treated patients in both studies (). In contrast, the responder rate was greater with PER 8 mg/day and PER 12 mg/day than with placebo in study 305 but did not significantly differ across groups in study 304 (). When both PER arms were pooled together and compared to placebo, the relative risk of the responder rate was 1.39 (95% CI 0.99–1.94, P = 0.06) in study 304 and 3.81 (95% CI 2.48–5.85, P < 0.001) (). Among the patients who completed study 304, none of those assigned to placebo were seizure free during the entire maintenance period in comparison with 2.2% with PER 8 mg/day and 1.5% with PER 12 mg/day. In study 305, the results were comparable with a seizure-free rate of 1.5% with placebo, 2.3% with PER 8 mg/day and 5% with PER 12 mg/day.
Figure 1 Clinical efficacy of PER in Phase III regulatory trials.
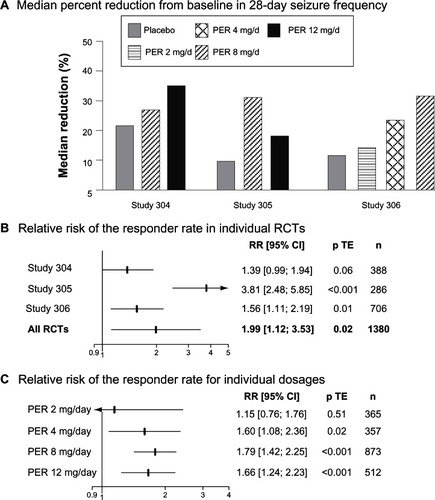
Study 306 included 706 patients.Citation28 180 were assigned to once-daily PER 2 mg/day, 172 to PER 4 mg/day, 169 to PER 8 mg/day and 184 to placebo. Patients entered a 6-week prospective baseline period before following a 19-week double-blind treatment phase that consisted of 6-week titration and 13-week maintenance periods. Baseline patients’ characteristics were comparable between treatment groups. However, it should be noted that the proportion of non-Caucasian patients was greater in study 306 (35%) than in studies 304 (13.9%) and 305 (16.6%). Patients were on average 33 years of age (range 12–72 years). The mean number of years since diagnosis of epilepsy was 19.1. Despite 1 to 3 AEDs, the baseline median seizure frequency per 28 days ranged from 9.3 to 10.9. Median reduction in seizure frequency was significantly greater with both PER 4 mg/day (23.3%) and PER 8 mg/day (30.8%) than with placebo (10.7%). In contrast, PER 2 mg/day (13.6%) did not prove superior to placebo. Similarly, the responder rate in PER groups increased in a dose-dependent manner from 20.6% with PER 2 mg/day, to 28.5% with PER mg/day and 34.9% with PER 8 mg/day, with only the responder rates observed with the two highest PER doses being significantly different from that observed in the placebo group (17.9%). Among the patients who completed the maintenance period, those seizure-free during the entire maintenance period were 1.2% in the placebo group, 1.9% in PER 2 mg/day, 4.4% in PER 4 mg/day and 4.8% in PER 8 mg/day.
Overall, these three Phase-III RCTs demonstrated the clinical efficacy of adjunctive once-daily PER 4 mg/day, 8 mg/day and 12 mg/day in adult drug-resistant partial epilepsy (). In contrast, PER 2 mg/day did not significantly differ from placebo. Surprisingly, although study 306 showed a dose response in seizure reduction, this observation was not confirmed when the data of the three studies were pooled together. Indeed, the CIs of the relative risks of the responder rates overlapped for all doses (). This lack of significant dose-response relationship might reflect methodological issues, including the large variability in the response to placebo. Indeed, the responder rate in the placebo group varied from 14.7% in study 305 to 26.4% in study 304. The response to placebo in study 304 was thus in the highest range in comparison with that previously observed in adult adjunctive-therapy partial epilepsy RCTs.Citation31,Citation33 Interestingly, post-hoc analyses of study 304 showed that the responder rate with both placebo and PER greatly varied across the geographical location of the patients.Citation27 The response to placebo, PER 8 mg/day and PER 12 mg/day was 21.9%, 40.5% and 40.0% in North American patients (n = 227), but 33.3%, 33.9% and 30.2% in Central and South American patients (n = 160). The basis for the regional variation in responses was unexplained though the authors suggested that patient selection or study conduct might have been an issue in Central and South American centers.Citation27 These methodological issues also hampered the relevance of indirect comparisons of the clinical efficacy of PER with other AEDs. It has been shown that the progressive increase in placebo response over time was an important limiting factor for the interpretability of meta-analyses and indirect comparisons of AEDs in the adjunctive-therapy of refractory partial epilepsy.Citation31 The great variability of the response to placebo in the PER Phase-III RCTs reinforced this conclusion.
Long-term studies
Although these data remain to be confirmed in the larger population with extended follow-up, the long-term efficacy data currently available showed that the level of seizure control observed in short-term Phase II and Phase III RCTs is maintained with long-term treatment.
138 patients who completed Phase II studies 206 and 208 were included in an open-label extension study.Citation30 The mean ± standard deviation (SD) PER dose during the study was 7.3 ± 3.3 mg/day. The responder rates were 43.8%, 51.5%, 49%, and 50% in patients with at least 1 (n = 89), 2 (n = 66), 3 (n = 52), and 4 (n = 18) years exposure, respectively. Only four patients (2.9%) were seizure free.
1218 patients who completed Phase III studies 304, 305 and 306 were enrolled in an open-label extension study, whose interim results were recently published.Citation29 At the interim data cutoff, 840 patients remained on PER treatment whereas 356 (29.2%) had discontinued. The primary reason for discontinuation was adverse events in 125 patients (10.5%), subject choice in 107 (9.0%) and inadequate therapeutic effect in 88 (7.4%). The mean ± SD dose of PER across the entire extension treatment phase was 10.1 ± 2.3 mg/day. In this interim analysis, only 588 patients had at least 1 year exposure to PER and 19 at least 2 years exposure. The responder rate at the end of 1 year was 47.6% and 63.2% at the end of 2 years. However, it must be stressed that results from open-label extension studies typically use a denominator that does not reflect the progressive drop-out of nonresponders (eg, patients who completed 12 months of treatment), and thus overestimate the true rate of responders. At one year, 7.1% of patients were seizure free for the entire 12-month period.
Safety and tolerability
As for the other AEDs, the most common adverse events (AEs) associated with PER are nonspecific central nervous system (CNS) side effects (). Dizziness and somnolence were the most frequent AEs observed during the regulatory trials. The tolerability of PER was primarily determined by the daily dosage and influenced by the management of titration. In randomized placebo-controlled trials, the relative risk (95% CI) of the withdrawal rate for adverse events was 1.76 (0.71–4.37) for PER 2 mg/day, 0.77 (0.25–2.38) for PER 4 mg/day, 1.59 (0.93–2.71) for PER 8 mg/d and 3.36 (1.96–6.11) for PER 12 mg/day (). PER 12 mg/day was thus the only dose associated with a significant greater withdrawal rate for adverse events than placebo. Some potentially severe adverse events were also more frequently observed with PER 12 mg/day, including falling over, which occurred in 10% of patients randomized to PER 12 mg/day in comparison with 3.4% in those randomized to placebo. This increased rate in falls with higher doses of PER has been related to the high incidence of somnolence and dizziness, leading to a specific alert about this issue in the European Summary of Product Characteristics.Citation23 Another warning was related to the use of PER in patients with a history of psychotic disorders. Indeed, 15.3% of patients exposed to PER during Phase III RCTs experienced psychiatric disorders, primarily insomnia, anxiety and aggressivity. In addition, eight cases of psychotic disorders and two cases of acute psychosis occurred in PER treated patients in comparison with respectively one and none for patients assigned to placebo.Citation23
Figure 2 Tolerability of PER in Phase III regulatory trials.
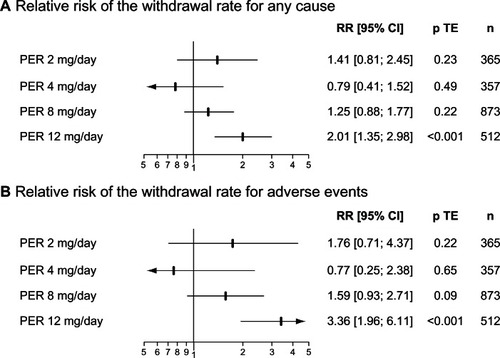
Table 3 Adverse events reported with PER in Phase III regulatory trials
Weight increase > 7% was observed in 19.2% of PER treated patients without apparent dose effect in study 304, in comparison with 8.3% in the placebo group.Citation27 This observation was confirmed in long-term extension studies where a clinically notable increase in body weight (>7%) was experienced by 29.5% of patients.Citation29
No evidence of an adverse effect of PER either on the hepatobiliary function, the renal function or shown by electrocardiography emerged. No death occurred during Phase II and Phase III studies. One death, related to possible Sudden Unexplained Death in Epilepsy (SUDEP), occurred in the extension phase of Phase II studies.Citation30 Three deaths occurred during the extension phase of Phase III studies. One was due to SUDEP, another to cerebral hemorrhage and the last to a road traffic accident.Citation29
According to the abuse potential of other glutamatergic neurotransmission antagonists such as ketamine, some concerns were raised about the issue of drug abuse and dependency with PER. A study examining the effects of doses up to 36 mg/day of PER in comparison with alprazolam and ketamine was conducted in recreational polydrug users.Citation23 PER could produce euphoric symptoms at doses of 24 or 36 mg/day. However, these effects were comparable to alprazolam, both in magnitude of effect, onset of action, and duration of effect. In contrast, positive subjective effects were lower than those produced by ketamine. Overall, the European Medicines Agency has estimated that the risk of PER abuse and dependency is limited, especially in the population of patients with epilepsy, and similar to that of alprazolam.Citation23
Conclusion
PER is a novel AED that exhibits a new mechanism of action: decreasing neuronal excitability by impacting glutamatergic transmission through noncompetitive blockade of AMPA receptors. In placebo-controlled trials in adults with refractory partial epilepsy, PER significantly reduced partial seizure frequency and was well tolerated. The main safety issues relate to dose-dependent nonspecific CNS-related adverse events, to potential risk of clinically notable weight gain and to psychiatric adverse events. However, in the absence of a direct comparison between PER and other licensed AEDs’ efficacy and tolerability, the clinical advantages of PER over the other AEDs in intractable partial epilepsy remain to be determined. Similarly, whether combining treatment of PER with other AEDs may have a synergistic effect on the reduction of neuronal hyperexcitability remains an open question. Although PER showed a significant antiepileptic efficacy in patients with refractory partial epilepsy included in Phase III studies and their open-label extension phase, only a minority of them achieved seizure freedom, a result similar to those previously reported over the last 20 years with other AEDs in similar populations.Citation2,Citation3,Citation34 In this context, it might be argued that the probability of PER having a major impact on a significant proportion of refractory patients remains low. Still, PER may help to reduce the frequency and severity of seizures in a significant number of patients with refractory epilepsy and, as suggested previously, may contribute to the reduction in the risk of sudden unexpected death in epilepsy in this highly exposed population.Citation35
Disclosure
Sylvain Rheims has received speaker fees from GSK and UCB Pharma and EISAI. Philippe Ryvlin has received speaker or consultant fees from Pfizer, Sanofi-Aventis, GSK, Jansen-Cilag, UCB Pharma, EISAI and Valeant.
References
- PeruccaETomsonTThe pharmacological treatment of epilepsy in adultsLancet Neurol201110544645621511198
- KwanPBrodieMJEarly identification of refractory epilepsyN Engl J Med2000342531431910660394
- BrodieMJBarrySJBamagousGAPatterns of treatment response in newly diagnosed epilepsyNeurol2012782015481554
- PeruccaPGilliamFGAdverse effects of antiepileptic drugsLancet Neurol201211979280222832500
- LoscherWMechanisms of drug resistanceEpileptic Disord20057Suppl 1S3S916120487
- PitkanenALukasiukKMechanisms of epileptogenesis and potential treatment targetsLancet Neurol201110217318621256455
- WhiteHSSmithMDWilcoxKSMechanisms of action of antiepileptic drugsInt Rev Neurobiol2007818511017433919
- StephenLJBrodieMJAntiepileptic drug monotherapy versus polytherapy: pursuing seizure freedom and tolerability in adultsCurr Opin Neurol201225216417222322411
- MayerMLArmstrongNStructure and function of glutamate receptor ion channelsAnnu Rev Physiol20046616118114977400
- HwaGGAvoliMThe involvement of excitatory amino acids in neocortical epileptogenesis: NMDA and non-NMDA receptorsExp Brain Res19918622482561684548
- LeeWLHablitzJJInvolvement of non-NMDA receptors in picrotoxin-induced epileptiform activity in the hippocampusNeurosci Lett19891071–31291342575723
- PerreaultPAvoliMPhysiology and pharmacology of epileptiform activity induced by 4-aminopyridine in rat hippocampal slicesJ Neurophysiol19916547717851675671
- BarnaBSzaszAVilagiIAnticonvulsive effect of AMPA receptor antagonist GYKI 52466 on 4-aminopyridine-induced cortical ictal activity in ratBrain Res Bull200051324124810718516
- NeumanRCherubiniEBen-AriYEpileptiform bursts elicited in CA3 hippocampal neurons by a variety of convulsants are not blocked by N-methyl-D-aspartate antagonistsBrain Res198845922652742902900
- HwaGGAvoliMExcitatory synaptic transmission mediated by NMDA and non-NMDA receptors in the superficial/middle layers of the epileptogenic human neocortex maintained in vitroNeurosci Lett19921431–283861359479
- RogawskiMARevisiting AMPA receptors as an antiepileptic drug targetEpilepsy Curr2011112566321686307
- ChappellASSanderJWBrodieMJA crossover, add-on trial of talampanel in patients with refractory partial seizuresNeurol2002581116801682
- LanganYMLucasRJewellHTalampanel, a new antiepileptic drug: single- and multiple-dose pharmacokinetics and initial 1-week experience in patients with chronic intractable epilepsyEpilepsia2003441465312581229
- HanadaTHashizumeYTokuharaNPerampanel: a novel, orally active, noncompetitive AMPA-receptor antagonist that reduces seizure activity in rodent models of epilepsyEpilepsia20115271331134021635236
- CeolinLBortolottoZABannisterNA novel anti-epileptic agent, perampanel, selectively inhibits AMPA receptor-mediated synaptic transmission in the hippocampusNeurochem Int201261451752222433907
- BalannikVMennitiFSPaternainAVMolecular mechanism of AMPA receptor noncompetitive antagonismNeuron200548227928816242408
- TempletonDFerryJPharmacokinetics of perampanel, a highly selective AMPA-type glutamate receptor antagonistEpilepsia2009509899
- Fycompa [perampanel] [webpage on the Internet]European Medicines Agency2012 [updated November 27, 2012]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002434/human_med_001572.jsp&mid=WC0b01ac058001d124Accessed March 1, 2013
- MarsonAGWilliamsonPRInterpreting regulatory trials in epilepsyCurr Opin Neurol200922216717319532041
- KraussGLBarMBitonVTolerability and safety of perampanel: two randomized dose-escalation studiesActa Neurol Scand2012125181521883097
- FrenchJAKraussGLSteinhoffBJEvaluation of adjunctive perampanel in patients with refractory partial-onset seizures: results of randomized global phase III study 305Epilepsia201354111712522905857
- FrenchJAKraussGLBitonVAdjunctive perampanel for refractory partial-onset seizures: randomized phase III study 304Neurol2012796589596
- KraussGLSerratosaJMVillanuevaVRandomized phase III study 306: adjunctive perampanel for refractory partial-onset seizuresNeurol2012781814081415
- KraussGLPeruccaEBen-MenachemEPerampanel, a selective, noncompetitive alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor antagonist, as adjunctive therapy for refractory partial-onset seizures: interim results from phase III, extension study 307Epilepsia201354112613422905878
- RektorIKraussGLBarMPerampanel Study 207: long-term open-label evaluation in patients with epilepsyActa Neurol Scand2012126426326922913800
- RheimsSPeruccaECucheratMFactors determining response to antiepileptic drugs in randomized controlled trials. A systematic review and meta-analysisEpilepsia201152221923321269281
- GazzolaDMBalcerLJFrenchJASeizure-free outcome in randomized add-on trials of the new antiepileptic drugsEpilepsia20074871303130717521343
- RheimsSCucheratMArzimanoglouAGreater response to placebo in children than in adults: a systematic review and meta-analysis in drug-resistant partial epilepsyPLoS Med200858e16618700812
- CallaghanBCAnandKHesdorfferDLikelihood of seizure remission in an adult population with refractory epilepsyAnn Neurol200762438238917880009
- RyvlinPCucheratMRheimsSRisk of sudden unexpected death in epilepsy in patients given adjunctive antiepileptic treatment for refractory seizures: a meta-analysis of placebo-controlled randomised trialsLancet Neurol2011101196196821937278