Abstract
The 22q13.3 deletion, or Phelan-McDermid syndrome, is characterized by global intellectual disability, generalized hypotonia, severely delayed or absent speech associated with features of autism spectrum disorder, and minor dysmorphisms. Its behavioral phenotype comprises sleep disturbances, communication deficits, and motor perseverations. Data on psychological dysfunctions are so far not available. Previous studies have suggested that the loss of one copy of the gene SH3 and multiple ankyrin repeat domains 3 (SHANK3) is related to the neurobehavioral phenotype. Additional genes proximal to SHANK3 are also likely to play a role in the phenotype of patients with larger deletions. The present paper describes two adult brothers with an identical 2.15 Mb 22qter (22q13.32q13.33) deletion, of whom the youngest was referred for evaluation of recurrent mood changes. In both patients, magnetic resonance imaging of the brain showed hypoplasia of the vermis cerebelli. Extensive clinical examinations led to a final diagnosis of atypical bipolar disorder, of which symptoms fully remitted during treatment with a mood stabilizer. In the older brother, a similar psychopathological picture appeared to be present, although less severe and with a later onset. It is concluded that the behavioral phenotype of the 22q13.3 deletion syndrome comprises absent or delayed speech and perseverations with associated autistic-like features, whereas its psychopathological phenotype comprises an atypical bipolar disorder. The latter may have implications for the treatment regime of the syndrome-related behavioral disturbances.
Introduction
More than a decade ago, a small number of patients were described in whom a telomeric deletion of the chromosomal region 22q13 was present and whose main phenotypical characteristics included marked developmental delay and absence of speech.Citation1–Citation3 Subsequently, the clinical picture of the 22q13 deletion syndrome was systematically elaborated by Phelan et al.Citation4 According to their report, this deletion syndrome is characterized by global intellectual disability (ID), childhood hypotonia, severely delayed or absent speech, features of autism spectrum disorder (ASD) and normal to accelerated growth in the absence of major dysmorphisms and somatic anomalies. Since then, the 22q13 deletion syndrome has also been called Phelan-McDermid syndrome (OMIM: 606232), and more than 100 cases with deletions of variable length have been published.Citation5–Citation7 The exact prevalence is unknown and the syndrome is thought to be underdiagnosed. In a large cohort study among 11,000 patients with ID, it was the second most frequent subtelomeric rearrangement, suggesting that it is a relatively common chromosomal disorder.Citation5
The SH3 and multiple ankyrin repeat domains 3 (SHANK3) gene that is localized within the common deleted region were suggested to be responsible for at least the neurobehavioral features, including ID and features of ASD.Citation8,Citation9 This was supported by the presence of a balanced translocation disrupting SHANK3 in a patient with the 22q13 deletion syndrome phenotype,Citation10,Citation11 and further confirmed by patients with deletions restricted to SHANK3. The latter is a brain-specific gene and has a critical function for the postsynaptic densities in the brain. Several previous studies suggested involvement of this gene in ASD.Citation12–Citation16 Recently, Sarasua et alCitation7 showed that several features of 22q13 deletion syndrome, including hypotonia and severity of ID, correspond with the deletion size, suggesting that additional genes proximal to SHANK3 contribute to the phenotype of these patients as well.
Concerning the behavioral phenotype, several reports mention sleep disturbances, communication deficits, and behavioral abnormalities such as hyperactivity, restlessness, attention problems, repetitive motor activities, and increased reactivity to sensory stimuli. Features concerning language and social communication are typically subsumed under the heading of ASD.Citation12,Citation13,Citation16–Citation19 Apart from the developmental aspects mentioned here, data about psychological dysfunctions or psychiatric symptoms are not available as yet.
In an ongoing cohort study among patients with unexplained ID (Department of Human Genetics, Radboud University Nijmegen Medical Centre, Nijmegen, The Netherlands), genome-wide array analysis disclosed a 2.15 Mb identical 22qter deletion (22q13.32q13.33; ) in two adult male siblings of whom the youngest was already known (first author) with an unstable mood pattern. In the absence of any relevant information about psychological dysfunctioning in this deletion syndrome, both patients were extensively examined in order to identify a possible psychopathological phenotype.
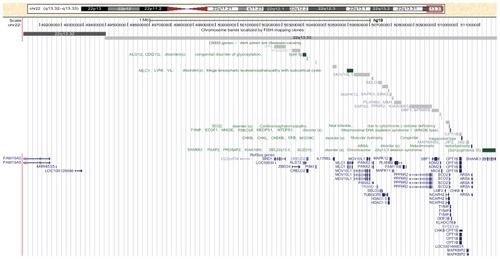
Figure 1 University of California, Santa Cruz genome browser view (GRCh37/hg19) of the deleted region in chromosome 22q13 identified by genome-wide array analysis.
Cases
Two male siblings are presented, aged 29 years (Patient 1) and 31 years (Patient 2), respectively, both born after uncomplicated pregnancies from nonconsanguineous parents. Apart from short-lasting episodes of flattened affect in the father, family history shows no load with any major psychiatric disorder, nor with ID or epilepsy.
Patient 1
The early childhood of this patient was characterized by sleep disturbances and a general developmental delay, particularly in respect of speech. He frequently displayed hyperactive behaviors with temper tantrums. At the age of 6 years he had a developmental delay of more than 3 years (developmental age 2 years 8 months). Aged 9 years, his IQ level corresponded with severe ID. Because of this and his recurrent challenging behaviors, the patient had special education since the age of 9 years. His medical history mentioned only bilateral orchidopexy at the age of 11 years. Aged 17 years, marked weight loss occurred and several changes in his behavioral repertoire became apparent, in that he showed a nearly complete loss of interest in daily activities, with marked social withdrawal as well as severe anxieties. A provisional diagnosis of major depression was made and the patient was treated with fluoxetine. However, that had to be stopped after about 6 months because of the occurrence of behavioral side effects, particularly impulsivities. Two years later, disinhibited behaviors, sleep disturbances, and obsessive rituals intensified. Genetic analysis at that time showed a normal 46,XY karyotype, and Fragile-X syndrome was excluded by FMR1 repeat analysis. Again, depression was thought to be the primary cause of the behavioral problems and, consequently, treatment with citalopram was started, which because of persistent motor agitation was replaced some months later by valproic acid in combination with haloperidol. Despite maintenance therapy with valproic acid (900–1200 mg daily) over a period of 4 years, he relapsed and was referred to the specialized outpatient department of neuropsychiatry.
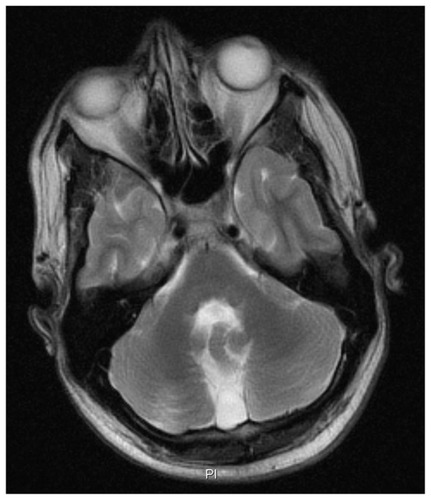
Psychiatric examination disclosed symptoms of a severe major depression with irritability, loss of initiative, and marked sleep disturbances. Magnetic resonance imaging (MRI) of the brain showed hypoplasia of the cerebellar vermis with enlarged cisterna magna, and mild enlargement of the lateral ventricles (). Neuropsychological assessment revealed severe ID (Vineland screener: 2 years 4 months) with marked deficits in expressive and receptive language. His cognitive profile was dominated by disordered attention and executive functioning, specifically expressed in hardly uninterruptable perseverative behaviors. Citalopram was reintroduced in an increasing dose of 40 mg daily, but in the absence of effect was replaced 3 months later by nortriptyline (50 mg daily; plasma concentration 176 μg/L). Subsequent CYP2D6 genotyping revealed a polymorphism (*4/*41; intermediate metabolism) and, based on the plasma concentration, the daily dose of nortriptyline was fixed at 40 mg daily (plasma concentration: 96 μg/L). Valproic acid was continued at a stable dose of 1200 mg per day (plasma concentration: 58 mg/L). A definite diagnosis of atypical bipolar disorder was made (International Classification of Diseases-10; F.31.9). Follow-up during a period of 5 years showed that his mood and behavior clearly stabilized.
Figure 2 Transversal T2-weighted magnetic resonance imaging of the brain of Patient 1, showing hypoplasia of the cerebellar vermis.
Patient 2
Patient 2 is the older brother. His recent history mentioned an unstable pattern of mood and activity with predominant recurrent depressive episodes starting at the age of 27 years for which he was treated with carbamazepine (400 mg daily; 4.5 mg/L) and paroxetine (30 mg daily; 39 μg/L) elsewhere. Four years later, after establishment of his genetic diagnosis, he was referred for extensive neuropsychiatric evaluation, which did not disclose any psychopathology. In respect of neuropsychology, a developmental age of 3 years 3 months (Vineland screener) was established, with a corresponding cognitive profile and communication problems due to moderately impaired development of speech and language. In particular, deficits in planning and maintaining overview were found. MRI of the brain disclosed a similar picture as in his younger brother (hypoplasia of the vermis cerebelli with enlarged cisterna magna, and mild enlargement of the lateral ventricles). Also here, in retrospect, a diagnosis of atypical bipolar disorder was made, albeit that relapsing symptomatology was less severe and with a later onset. Because psychiatric symptoms were in full remission, his psychopharmacological treatment regime was kept unchanged.
As shown in , primary behavioral and psychopathological features of both patients are from the mood and anxiety domains.
Table 1 Behavioral and psychopathological findings in both patients
Genome-wide submicroscopic chromosomal analysis by 250 k single nucleotide polymorphism array analysis (Affymetrix, Inc, Santa Clara, CA) revealed in both brothers an identical terminal loss of 2.15 Mb in chromosomal region 22q13.32q13.33 (49.03–51.18 Mb according to University of California, Santa Cruz genome browser Hg 19). Chromosomal analysis in the parents gave normal results without any indications for a mosaic pattern (blood lymphocytes and buccal cells).
Discussion
In these brothers, with until recently unknown etiology of their severe ID and moderate to marked developmental delay of speech and language, genome-wide array analysis revealed a diagnosis of 22q13 deletion syndrome/Phelan-McDermid syndrome, which appeared to be associated with a long-lasting pattern of unstable mood and behavior. In the absence of a family load with (bipolar) affective disorder, in both brothers a psychiatric diagnosis of atypical bipolar disorder was made, albeit that the youngest patient presented with more pronounced symptoms and an onset at earlier age. Their respective psychiatric histories disclosed no symptoms belonging to the autism spectrum.
Although several authors have suggested that autistic symptoms belong to the behavioral phenotype,Citation4,Citation12,Citation16–Citation18,Citation20,Citation21 the incremental value of such a diagnostic vignette is only marginal, given the co-occurrence with severe ID and, as was also recently stressed by Sarasua et alCitation7, relies heavily on the definition and the validity of the assessment process. In these patients, it is not autism that is at the heart of the phenotypical presentation but the characteristic combination of marked speech and language delay and executive dysfunctions, the latter mainly detectable through a pattern of perseverations and planning problems.
In retrospect, it can be argued that the fundamental lack of expressive language not only intensified the presentation of previously observed symptoms of major depression but also obscured the fluctuations in mood and behavior typical for so-called unstable mood disorder.Citation22 This psychopathological syndrome that refers to an atypical bipolar disorder can frequently be observed in patients with ID irrespective of the etiology.Citation23,Citation24 The dysregulation of affect in these patients is most probably related to hypoplasia of the cerebellar vermis that is typically involved in the modulation of cognition and emotion.Citation25,Citation26
As their symptoms remitted under maintenance therapy with a mood stabilizer, compounds from this class may be the first choice in the treatment of fluctuating challenging behaviors in patients with ID and, more specifically, those with 22q13 deletion syndrome in whom an (atypical) bipolar disorder may be present. These patients also illustrate that categorical diagnoses of autism or ASD should be applied with great caution only. Such vignettes may seriously hinder the in-depth assessment of an individual patient’s functioning following the contextual neuropsychological paradigm.Citation27 The latter is aimed to understand and to explain functional deficits and is relevant for the treatment design.
In conclusion, key elements of the behavioral phenotype of the 22q13.3 deletion syndrome, as previously described in the literature, are delayed or absent speech and perseverations with associated features from the autism spectrum. This is the first report of which the data suggest that the psychopathological phenotype of Phelan-McDermid syndrome comprises oscillating abnormalities in mood and behavior that can be attributed to an atypical bipolar disorder.
Acknowledgments
This study is part of a collaborative project of the research group Psychopathology and Genetics of the Radboud University Nijmegen and the Vincent van Gogh Institute for Psychiatry, Venray, The Netherlands. The authors are indebted to the patients and their parents for their cooperation and to Bart PC van de Warrenburg, a neurologist at the Department of Neurology, Radboud University Medical Centre Nijmegen, for the interpretation of the brain magnetic resonance imaging. Written informed consent was obtained from the parents for publication of the case reports.
Disclosure
The authors report no conflicts of interest in this work.
References
- FlintJWilkieAOBuckleVJWinterRMHollandAJMcDermidHEThe detection of subtelomeric chromosomal rearrangements in idiopathic mental retardationNat Genet199591321407719339
- WongACCNingYFlintJMolecular characterization of a 130-kb terminal microdeletion at 22q in a child with mild mental retardationAm J Hum Genet1997601131208981954
- PrechtKSLeseCMSpiroRPTwo 22q telomere deletions serendipitously detected by FISHJ Med Genet1998359399429832042
- PhelanMCRogersRCSaulRA22q13 Deletion syndromeAm J Med Genet2001101919911391650
- RavnanJBTepperbergJHPapenhausenPSubtelomere FISH analysis of 11688 cases: an evaluation of the frequency and pattern of subtelomere rearrangements in individuals with developmental disabilitiesJ Med Genet20064347848916199540
- BonagliaMCGiordaRBeriSMolecular mechanisms generating and stabilizing terminal 22q13 deletions in 44 subjects with Phelan/McDermid syndromePloS Genetics20117e100217321779178
- SarasuaSMDwivideABoccutoLAssociation between deletion size and important phenotypes expands the genomic region of interest in Phelan-McDermid syndrome (22q13 deletion syndrome)J Med Genet20114876176621984749
- WilsonHLCrollaJAWalkerDIntersitial 22q13 deletions: genes other than SHANK3 have major effects on cognitive and language developmentEur J Hum Genet2003161301131018523453
- PhelanMCDeletion 22q13.3 syndromeOrph J Rare Dis2008314
- BonagliaMCGiordaRBorgattiRDisruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndromeAm J Hum Genet20016926126811431708
- BonagliaMCGiordaRManiEIdentification of a recurrent breakpoint within the SHANK3 gene in the 22q13.3 deletion syndromeJ Med Genet20064382282816284256
- PrasadCPrasadANChodirkerBNGenetic evaluation of pervasive developmental disorders: the terminal 22q13 deletion syndrome may represent a recognizable phenotypeClin Genet20005710310910735630
- LucianiJJde MasPDepetrisDTelomeric 22q13 deletions resulting from rings, simple deletions and translocations: cytogenetic, molecular and clinical analyses of 32 new observationsJ Med Genet20034069069612960216
- MoessnerRMarshallCRSutcliffeJContribution of SHANK3 mutations to autism spectrum disorderAm J Med Genet20078112981297
- DurandCMBetancurCBoeckersTMMutations in de gene encoding the synaptic scafolding protein SHANK3 are associated with autism spectrum disordersNat Genet200739252717173049
- PhilippeABoddaertNVaivre-DouretLNeurobehavioral profile and brain imaging study of the 22q13.3 deletion syndrome in childhoodPediatrics2008122376382
- HavensJMVisootsakJPhelanMCGrahamJM22q13 deletion syndrome: an update and review for the primary pediatricianClin Pediatr2004434353
- Cusmano-OzogKManningMAHoymeHE22q13.3 deletion syndrome: a recognizable malformation syndrome associated with marked speech and language delayAm J Med Genet2007145C39339817926345
- DharSUDel-GaudioDGermanJR22q13.3 Deletion syndrome: clinical and molecular analysis using array CGHAm J Med Genet2010152A57358120186804
- ManningMACassidySBClericuzioCTerminal 22q deletion syndrome: a newly recognized cause of speech and language disability in the autism spectrumPediatrics200411445145715286229
- LindquistSGKirchhoffMLundsteenCFurther deleneation of the 22q13 syndromeClin Dysmorphol200514556015770125
- VerhoevenWMATuinierSCyclothymia or unstable mood disorder. A systematic treatment evaluation with valproic acidJ Appl Res Intellect Disabil20011410471154
- GualtieriCTBrain injury and mental retardation: psychopharmacology and neuropsychiatryPhiladelphia, PALippincott Williams & Wilkins2002
- VerhoevenWMASijbenAESTuinierSPsychiatric consultation in intellectual disability: demensions, domains and vulnerabilityEur J Psychiat2004183143
- SchmahmannJDWeilberJBShermanJCThe neuropsychiatry of the cerebellum: insights from the clinicCerebellum2007625426717786822
- HoppenbrouwersSSSchutterDJLGFitzgeraldPBChenRDaskalakisZJThe role of the cerebellum in the pathophysiology and treatment of neuropsychiatric disoders: a reviewBrain Res Rev20085918520018687358
- EggerJIMDe MeyHRAJanssenGAssessment of executive functioning in psychiatric disorders: functional diagnosis as the ouverture of treatmentClin Neuropsychiatr20074111116
- Van DuinGDijkshoornYNoensIScholteEvan Berckelaer-OnnesIVineland screener 0–12 years research version (NL). Constructing a screening instrument to assess adaptive behaviourInt J Methods Psychiatr Res20091811011719507164
- MatsonJLKazdinAESenatoreVPsychometric properties of the Psychopathological Instrument for Mentally Retarded AdultsAppl Res Ment Retard1984581896721483
- CummingsJLMegaMGrayKThe Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementiaNeurology199444230823147991117