
Abstract
Background
The ketogenic diet (KD) is a proven therapy for refractory epilepsy. Although the anti-seizure properties of this diet are understood to a certain extent, the exploration of its neuroprotective effects and underlying mechanisms is still in its infancy. Tissue acidosis is a common feature of epileptogenic foci. Interestingly, the activation of acid-sensing ion channel 1a (ASIC1a), which mediates Ca2+-dependent neuronal injury during acidosis, has been found to be inhibited by ketone bodies in vitro. This prompted us to investigate whether the neuroprotective effects induced by the KD occur via ASIC1a and interconnected downstream mechanisms in a rat model of temporal lobe epilepsy.
Methods
Male Sprague-Dawley rats were fed either the KD or a normal diet for four weeks after undergoing pilocarpine-induced status epilepticus (SE). The effects of KD on epileptogenesis, cognitive impairment and hippocampal neuron injury in the epileptic rats were subsequently evaluated by video electroencephalogram, Morris water maze test and Nissl staining, respectively. The expression of ASIC1a and cleaved caspase-3 in the hippocampus were determined using Western blot analysis during the chronic period following SE. Moreover, the intracellular Ca2+ concentration, mitochondrial membrane potential (MMP), mitochondrial reactive oxygen species (mROS) and cell apoptosis of hippocampal cells were detected by flow cytometry.
Results
We found that the KD treatment strongly attenuated the spontaneous recurrent seizures, ameliorated learning and memory impairments and prevented hippocampal neuronal injury and apoptosis. The KD was also shown to inhibit the upregulation of ASIC1a and the ensuing intracellular Ca2+ overload in the hippocampus of the epileptic rats. Furthermore, the seizure-induced structure disruption of neuronal mitochondria, loss of MMP and accumulation of mROS were reversed by the KD treatment, suggesting that it has protective effects on mitochondria. Finally, the activation of caspase-3 was also inhibited by the KD.
Conclusion
These findings indicate that the KD suppresses mitochondria-mediated apoptosis possibly by regulating ASIC1a to exert neuroprotective effects. This may provide a mechanistic explanation of the therapeutic effects of KD.
Introduction
Temporal lobe epilepsy (TLE), the most common form of chronic epilepsy, has a high rate of drug resistance as more than one-third of patients do not achieve good control with available drugs and eventually progresses to refractory epilepsy.Citation1 Cognitive decline is an increasingly recognised comorbidity of TLE, with a higher prevalence of dementia,Citation2,Citation3 which greatly diminishes one’s quality of life. Therefore, efforts aimed at antiepileptogenesis and improving cognitive function using an effective intervention are urgently needed.
The ketogenic diet (KD), a high-fat, low-carbohydrate and adequate-protein diet, is well known for its efficacy in treating drug-resistant epilepsies. In addition to its anti-seizure properties, evidence suggests that the KD may exert cognitive improvement and neuroprotective effects.Citation4–6 However, despite its clear clinical efficacy, its application is restricted due to poor dietary compliance and several adverse reactions. Therefore, understanding the underlying mechanisms of its therapeutic effects will not only provide insight into the mechanisms of epilepsy but also facilitate the development of effective pharmacological strategies. Previous studies have shown the effects of the KD on modulating neurotransmitter levels, anti-oxidative stress, maintaining energy metabolism and reducing inflammation,Citation5 but the exact mechanism underlying its neuroprotective effects remains poorly understood to date.
Hippocampal sclerosis (degeneration), mainly characterised by gliosis and pyramidal neuronal loss, is the most common histopathological hallmark of TLE and is thought to be responsible for epileptogenesis and seizure-related cognitive dysfunction.Citation7–9 Undoubtedly, aggressive prevention of neuronal death is warranted. Acid-sensing ion channels (ASICs) are proton-gated sodium-selective channels widely expressed in central and peripheral nervous systems.Citation10,Citation11 Acid-sensing ion channel 1a (ASIC1a), the major functional subtype of ASICs, also induces direct Ca2+ influxes, triggers a cascade of reactions and eventually causes neuronal injury and death during tissue acidosis.Citation12,Citation13 Since the intensive electrical activity of neurons during epileptic seizures leads to a significant drop in the extracellular pH of the brain tissue,Citation14,Citation15 ASIC1a has become an attractive epilepsy research target. There is evidence that ASIC1a polymorphism is associated with TLE in a Han Chinese population.Citation16 The relationship between ASIC1a and epilepsy has been further verified in animal experiments, revealing that ASIC1a plays a crucial role in the generation and maintenance of epileptic seizures, as well as related neuronal injury.Citation17–19 In addition, evidence indicates that a crosstalk between ASIC1a and the mitochondria plays an important role in acidosis-induced neuronal injury.Citation20,Citation21 Thus, we postulate that ASIC1a and mitochondria may serve as potential therapeutic targets for TLE and its related neurodegeneration. Interestingly, a recent study implies that ketone bodies, i.e. the products of KD metabolism, provide antiepileptic and neuroprotective effects via inhibiting the opening of ASIC1a in rat hippocampal excitatory neurons in vitro.Citation22 However, this still needs to be further explored in vivo.
The present study is designed to further explore the effects of the KD on hippocampal neurodegeneration and the possible involvement of ASIC1a and its related mitochondrial pathways in a pilocarpine rat model of TLE, which is considered a good model of TLE since it reproduces the main characteristics of human TLE, including delayed neuronal damage and epileptogenesis after status epilepticus (SE).Citation23 To better mimic the usual clinical situation, we initiate the KD post-treatment following pilocarpine-induced SE and subsequently evaluate its effects on epileptogenesis, cognitive function and hippocampal neuron apoptosis during the chronic period. Moreover, we attempt to demonstrate that these neuroprotective effects induced by the KD may involve ASIC1a and the mitochondria-mediated apoptosis pathway. This may provide a basis for identifying new therapeutic targets to halt hippocampal neurodegeneration and its related cognitive dysfunction in TLE.
Experimental Procedures
Animals
70 male Sprague-Dawley rats (6–8 weeks old, 180–200 g) were obtained from Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) [License no. SCXK (Jing) 2016-0006]. The animals were kept at 22°C ± 1°C under a normal 12-h light/dark cycle (lights on from 8:00 to 20:00), with free access to water and food. All experiments were approved by the Research Ethics Committee of the Second Hospital of Hebei Medical University and were performed in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Induction of SE
Persistent SE was induced by lithium-pilocarpine in the rats as described previously.Citation24 Briefly, 40 rats were intraperitoneally (i.p.) injected lithium chloride (127 mg/kg, Sigma, St. Louis, MO, USA) 18 h prior to a single injection of pilocarpine (50 mg/kg, i.p., P6503, Sigma, St. Louis, MO, USA). Atropine sulphate (1 mg/kg, i.p., Tianjin, China) was injected 30 min before the pilocarpine to reduce peripheral cholinergic effects. Seizures were graded using the Racine scale,Citation25 as follows stage 1, mouth and facial movements; stage 2, head nodding; stage 3, unilateral forelimb clonus; stage 4, rearing with bilateral forelimb clonus; and stage 5, rearing and falling. The beginning of SE was defined as a sustained seizure activity (≥stage 4). To terminate generalised convulsive seizures, repeated doses of chloral hydrate (300 mg/kg, i.p. Tianjin, China) were injected 60 min after the onset of SE. Eventually, 10 rats died after SE, 4 rats did not meet the criteria of SE, and the remaining 26 rats had SE successfully induced. The mortality during pilo-SE was 25%, and the rate of seizure development was 90%. Meanwhile, 30 rats as control group received the same dose of saline instead of pilocarpine.
Experimental Protocol and Diet Treatment
The remaining SE rats were randomly allocated into 2 groups: the Pilo group (n=13) that received the normal diet (ND), and the Pilo+KD group (n=13) that received the KD. The control rats were also randomly divided into 2 groups: the control group (n=13) that received the ND, and the KD alone group (n=13) that received the KD. A KD with a caloric composition of 10.4% protein, 89.5% fat and 0.1% carbohydrates was purchased from Shuyishuer Bio-tech Co., Ltd. (D12369B, Sysebio, Changzhou, China). The ND consisted of 23.1% protein, 11.8% fat, and 65.1% carbohydrates. The rats were kept in separate cages and fed ad libitum either the KD or ND for four weeks beginning three days after SE, similar to the previous studyCitation26 with slight modifications. In order to facilitate the development of ketonemia, the KD-fed rats were fasted overnight prior to the initiation of the diet treatment. The schematic design of this study is summarised in .
Figure 1 The β-hydroxybutyrate levels were measured with a ketone monitoring system. (A) Flow chart of the experimental procedures. Three days after SE, the rats were fed either the KD or a normal diet for 28 days. On the last day of the KD treatment, the rats’ ketone levels were measured and EEG electrodes were implanted. After a one-week post-surgery recovery period, continuous vEEG monitoring was used from day 38 to day 52 post-SE. An MWM test was conducted 6 days prior to sacrifice. (B) The β-hydroxybutyrate levels were measured with a ketone monitoring system after four weeks of KD treatment. (n=8 rats/group; one-way ANOVA with LSD test). The values were expressed as the mean ± SEM. ***Represents P < 0.001.
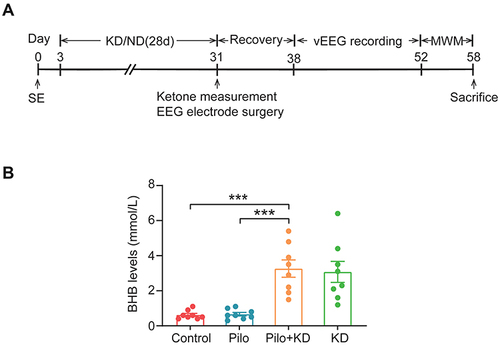
To ensure ketosis, the β-hydroxybutyrate levels were measured with a ketone monitoring system (FreeStyle Optium Neo, Abbott Diabetes Care Ltd., Oxon, UK) at 30 days post-SE. Blood was collected from the lateral tail vein.
Electrode Implantation and Video Electroencephalogram Recordings
Four weeks post SE, the rats were implanted with epidural electroencephalogram (EEG) electrodes. Briefly, the rats were fixed on a stereotaxic apparatus (RWD, Shenzhen, China) under anaesthesia, and a recording electrode made from stainless steel screws was placed 2 mm anterior and 2 mm lateral to the bregma. The reference and ground screw electrodes were positioned over the right and left occipital cortices, respectively. The electrodes were attached by a flexible wire to a micro connector and secured with dental acrylic. After a one-week post-surgery recovery period, the rats were individually placed in an acrylic cage and connected to an amplifier (Warner Instruments, USA) through a flexible cable. The EEG signals were recorded and digitised using a Power Lab 8/35 system (AD Instruments, Colorado Springs, CO, USA). Then continuous video electroencephalogram (vEEG) was recorded for up to 14 days. Generalised seizures were defined by stage 4 and 5 behavioral seizures, or repetitive epileptiform spiking activity (>5 Hz) that continued for >10s on the EEG.
The Morris Water Maze Test
The MWM test was conducted after the completion of the vEEG monitoring. The Morris water maze (MWM) apparatus consisted of a water tank (150 cm in diameter, 50 cm in height) that was filled with 22°C±1°C non-toxic black water and orientation was made possible by four visual cues placed on the surrounding walls. On the first day, a visible platform trial was conducted with a platform 1 cm above the surface of the water to habituate the rats to the testing environment. During the acquisition phase, the platform was submerged 1.5 cm below the water’s surface, the rats were put into the water at different start locations and had to swim freely to find the platform. The time each rat took to reach the platform (i.e. the escape latency) was recorded. Rats that failed to find the platform within 120 s were placed on the platform for 30s. The trials were performed four times a day for five consecutive days. On the last day, a probe trial was conducted. The platform was removed, and the rats were allowed to swim freely for 120s. The number of rats that crossed over the previous platform location and the time they spent in the previous platform’s quadrant were recorded and analysed.
Nissl Staining
After completing of the behavioural tests, the rats were anesthetised and transcardially perfused with cold saline, followed by 4% paraformaldehyde (PFA) in 0.1 M phosphate-buffered saline (PBS). The brains were removed and fixed in 4% PFA at 4°C for 24 h. The fixed specimens were embedded in paraffin and cut into 5-μm-thick sections. The coronal sections which come from 2 and 7mm posterior of the bregma and include the whole hippocampus were stained with 1% thionin as described previously.Citation27 The Nissl-stained slices (three slices per animal) were observed using a light microscope (Olympus BX51, Tokyo, Japan) at 200 magnification, and the number of the surviving pyramidal neurons per 1-mm2 area of hippocampal CA1 and CA3 subfields was counted in a blinded manner using the Image Pro Plus 6.0 software. 15 sections from three rats in each group were analyzed.
Transmission Electron Microscopy
The rats were anesthetised and transcardially perfused with cold saline, followed by 0.1 M PBS containing 4% PFA and 4% glutaraldehyde. The brains were removed, and 1-mm3 samples were taken from the hippocampal CA1 area. Tissue blocks were processed for electron microscopy according to the method described in a previous study.Citation28 The ultrastructural changes of the mitochondria were observed under a transmission electron microscope (JEM-1230; Jeol Ltd., Tokyo, Japan). Five fields of view were randomly selected at the same magnification, and a total of 50 mitochondria were analysed per animal. Mitochondrial damage was graded using Flameng’s scale,Citation29 as follows, 0 points normal structure; 1 point, matrix density reduced, cristae separated; 2 points, matrix is transparent, cristae separated but not broken; 3 points, matrix coagulation, cristae ruptured; and 4 points, the integrity of the inner and outer membranes disappears and becomes vacuolated.
Western Blotting
The hippocampus was freshly isolated, and the hippocampal protein was extracted using a commercial kit (Applygen Technologies, Beijing, China). The total protein concentrations were determined using a BCA protein assay kit (Beyotime, Shanghai, China). Equal amounts of protein were separated on 10% SDS-PAGE gels and transferred to immobilon PVDF membranes. After being blocked in 5% non-fat milk, the blots were incubated with primary antibodies against ASIC1a (1:1000, ab240896, Abcam), cleaved caspase-3 (1:500, BF0711, Affinity) and β-actin (1:10000, ab6276, Abcam) followed by horseradish peroxidase-conjugated secondary antibodies (1:3000, Elabscience). Finally, protein bands were visualised using an enhanced chemiluminescence method. The relative intensity of the target protein was normalised to β-actin and analysed using the ImageJ software.
Preparation of Single Cell Suspension for Flow Cytometry
The bilateral intact hippocampi were freshly isolated, rinsed with ice-cold PBS and minced with ophthalmic scissors. To obtain a single-cell suspension, the tissue was digested with 0.25% trypsin-EDTA at 37°C for 30 min and triturated with Pasteur pipettes. After being filtered through a 70-μm cell strainer, the suspension was collected and centrifuged at 1000 rpm for five minutes. The precipitated cells were resuspended in PBS.Citation30
Detection of Cell Apoptosis
An FITC AnnexinV apoptosis detection kit (BD Biosciences, San Jose, CA, USA) was used to detect cell apoptosis in the hippocampus. The single-cell suspension (at least 5000 cells from each sample) was incubated with 5 μL FITC Annexin V and 5 μL PI for 15 min at 25 °C in the dark. Subsequently, 400 μL of binding buffer was added to the stained cells, which were analysed by flow cytometry (CytomicsFC500, Beckman Coulter, USA). The data were analysed using the Expo32 ADC software.
Measurement of the Intracellular Ca2+ Concentration
To measure the intracellular Ca2+ concentration ([Ca2+]i) in the hippocampal cells, Fluo-3 AM (Invitrogen, Carlsbad, CA, USA) was used; it was added to the single-cell suspension (at least 5000 cells from each sample) to incubate for 30 min at 37°C away from light, washed with PBS, measured by flow cytometry at a 488-nm excitation wavelength and finally expressed as the relative fluorescence intensity.Citation31
Assessment of MMP
The MMP was assessed using a fluorescent JC-1 probe (Beyotime, Shanghai, China) according to the manufacturer’s protocol. The single-cell suspension (at least 5000 cells from each sample) was incubated with JC-1 working stock for 20 min at 37 °C in the dark, washed with JC-1 buffer solution, and then detected using flow cytometry.
Analysis of mROS
The mROS in the hippocampal cells (at least 5000 cells from each sample) were detected by DCFH-DA (Cayman, Ann Arbor, MI, USA) according to the manufacturer’s instructions. In brief, the cells were incubated with DCFH-DA (10 µM) at 37°C for 20–30 min, washed three times with PBS, measured by flow cytometry and finally expressed as the relative fluorescence intensity.
Statistical Analyses
All data were expressed as the mean ± SEM and checked using the Shapiro Wilk test before further analysis. The statistical significance between two groups was evaluated using the Student’s t-test. Four datasets were analysed with a one-way analysis of variance (ANOVA) (equal variance) followed by the least-significant difference (LSD) test or Kruskal Wallis test (unequal variance). Escape latencies in the MWM test were compared by a repeated-measures two-way ANOVA followed by the LSD test. The statistical analysis was performed using the SPSS 25.0 software (IBM, Armonk, NY, USA) and GraphPad Prism 9.0 software (GraphPad Software Inc., La Jolla, CA, USA). Finally, P-values <0.05 were regarded as statistically significant.
Results
The KD Attenuates Spontaneous Recurrent Seizures (SRSs) in the Chronic Period Following SE
The serum levels of β-hydroxybutyrate, a primary form of ketone, were significantly different among all of the groups (F(3, 28) = 13.700, P = 0.000) (). They were markedly elevated in the KD-fed rats compared with those in the ND-fed rats after four weeks of diet treatment (P < 0.001), indicating that the KD treatment successfully induced a state of ketosis.
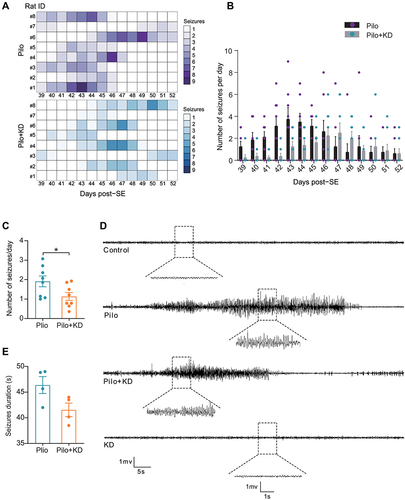
To test whether KD feeding prevents SE-induced SRSs, we used a continuous vEEG monitoring system from day 38 to day 52 post SE (). It revealed that all of the rats displayed SRSs, and seizure clusters were observed in several rats (). The number of seizures per day varied widely between rats, ranging from 0 to 9 in the Pilo group and ranging from 0 to 6 in the Pilo+KD group (). As expected, the frequency of SRSs was significantly decreased in the Pilo+KD group compared with the Pilo group (1.13 ± 0.21 seizures/day vs 1.91 ± 0.28 seizures/day, respectively; t(14) = 2.241, P = 0.042) (). Meanwhile, the Pilo group displayed robust prolonged seizures with a higher spike amplitude on the EEG, the seizure activity was attenuated after the KD treatment in the Pilo + KD group, as evidenced by the lower spike amplitude and shorter duration, although no statistical significance was found compared to the Pilo group (41.54 ± 1.32 s vs 46.38 ± 1.65 s, respectively; t(6) = 2.287, P = 0.062) ( and ). Neither seizures nor aberrant EEG activity were observed in the control group or KD alone group (). Together, these observations suggested that the KD exerts a suppressive effect on epileptogenesis.
Figure 2 Effects of the KD on SRSs assessed by continuous vEEG monitoring from day 38 to day 52 after lithium-pilocarpine induced SE. (A) The heat map of seizures occurring throughout the 14-day recording time for each individual rat. (B) The scatter plot of daily seizure frequency for each day during the recording time. (C) The effect of KD on the number of seizures per day was determined by behavioral observation. (n=8 rats/group; Student’s t test). (D) Representative EEG traces at the chronic phase after pilocarpine injection. (E) The effect of KD on the seizure duration was evaluated by EEG (n=4 rats/group; Student’s t test). The values were expressed as the mean ± SEM. *Represents P < 0.05.
The KD Ameliorates Learning and Memory Impairments in Rats with Chronic Seizures
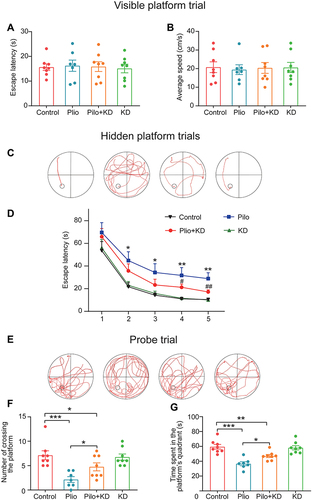
To evaluate the effects of the KD treatment on spatial learning and memory, we performed the MWM test (). There were no significant differences in escape latency (F (3, 27) = 0.065, P = 0.978) and swimming speed (F (3, 27) = 0.042, P = 0.988) among the four groups in the visible platform test, indicating no differential influences of vision and locomotor function on performance ( and ). During the five days of hidden platform trials, the swimming track was recorded (). As illustrated by , the performance of all four groups was significantly improved with training (group effect F (3, 120) = 11.222, P = 0.000; training day effect F (4, 117) = 30.430, P = 0.000). However, the rats in the Pilo group exhibited worse spatial learning manifesting as markedly prolonged escape latency compared to the rats in the control group (P<0.001). Interestingly, the Pilo+KD group showed significantly shorter latency time on days 4 and 5 compared to the Pilo group (P<0.01), suggesting that the KD treatment mitigated the poor performance of the epileptic rats. In the probe trial without the hidden platform, the swimming track was recorded, and significant differences were observed among all of the groups (F (3, 27) = 8.481, P = 0.000) (). Compared to the control group, the rats in the Pilo group showed poorer memory via fewer platform crossings (P = 0.000) and less time spent in the target quadrant (P = 0.000) ( and ). However, the rats in the Pilo+KD group exhibited a higher preference for the target quadrant (P = 0.012) and more platform crossings (P = 0.026) compared with the rats in the Pilo group but still less crossings than the rats in the control group. There were no significant differences in the learning and memory abilities between the control group and KD alone group (both P>0.05). These results indicated that the KD treatment ameliorates seizure-induced cognitive deficits.
Figure 3 Effects of the KD on learning and memory function in pilocarpine-treated rats as measured by MWM test. (A and B) The escape latency and average swimming speed were assessed in the visible platform trial. (C and D) The swimming track and the escape latency was recorded during the five days of hidden platform trials (*P < 0.01, **P < 0.001 vs the control group; #P < 0.05, ##P < 0.01 vs the Pilo group; repeated-measures ANOVA with LSD test). (E) The swimming track was recorded in a probe trial on the last day. (F and G) The number of crossings over the original platform location and the time spent in the platform quadrant were assessed in the probe trial. (*P < 0.05, **P < 0.01, ***P < 0.001; one-way ANOVA with LSD test). All data were presented as the mean ± SEM. Control (n=8 rats), Pilo (n=7 rats), Pilo+KD (n=8 rats) and KD (n=8 rats).
The KD Alleviates Seizure-Induced Neuronal Damage and Apoptosis in the Rat Hippocampus
Next, we set out to verify the protective effects of the KD on hippocampal neuron at 58 days post SE. Nissl staining was used to assess the pyramidal neuronal damage after the seizures (). It revealed that there were significant differences (CA1 F (3, 8) = 58.298, P = 0.000; CA3 F (3, 8) = 15.134, P = 0.001) in the number of surviving pyramidal neurons among the groups ( and ). And the number of surviving neurons in the hippocampal CA1 (P = 0.000) and CA3 (P = 0.001) regions was dramatically decreased in the Pilo group compared with the control group. Additionally, obvious pathological changes were observed in the hippocampal neurons of the epileptic rats, as manifested by light staining, loose and irregular arrangement, and the CA1 region being more severe than the CA3 region ( and ). However, the KD treatment markedly attenuated the pyramidal neuron loss (CA1 P = 0.003; CA3 P = 0.021) and reversed the morphological damage ( and ). There were no significant differences in the number or morphology of the surviving pyramidal neurons between the control group and KD alone group (CA1 P = 0.495; CA3 P = 0.784) ( and ).
Figure 4 Effects of the KD on hippocampal neuron damage and apoptosis in epileptic rats. The Nissl staining of the whole hippocampus (A–D) and CA1 subfield (E–H) and CA3 subfield (I–L). No neuron loss occurred in the hippocampus of the control group ((A), (E) and (I)) and KD alone group ((D), (H) and (L)). Marked neuron loss and injury in the CA1 and CA3 subfields of the hippocampus were observed in the Pilo group ((B), (F) and (J)). This neurodegeneration was effectively rescued by the KD treatment ((C), (G) and (K)). scale bar = 200 μm (A–D); scale bar = 50 μm (E–H) and (I–L). (M) The apoptotic cells in the hippocampus were measured by Annexin-V/PI staining and evaluated by flow cytometry. (N and O) Quantitative analysis of surviving pyramidal neurons per 1-mm2 area of CA1 and CA3 regions in the hippocampus (n = 3 rats/group). (P) Quantitative analysis results of the cell apoptosis rate (n = 4 rats/group). All results were expressed as the mean ± SEM, and analysed by a one-way ANOVA with an LSD test. *, **, ***Represent P < 0.05, P < 0.01 and P < 0.001, respectively.
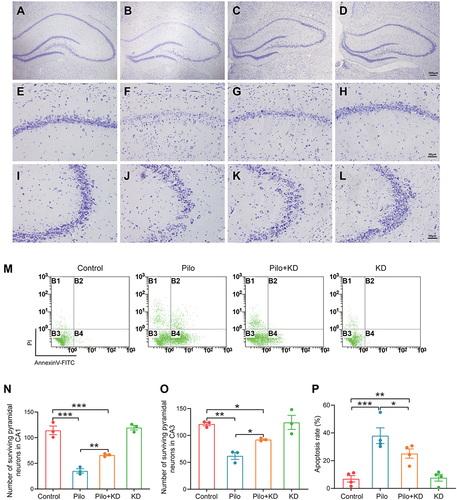
The apoptosis of the hippocampal cells was detected by flow cytometry with a combination of Annexin V and propidium iodide (PI) staining (). Annexin V can identify early apoptotic cells (B4, Annexin V+/PI−) via binding to cells with exposed phosphatidylserine, while PI can permeate into late apoptotic or necrotic cells (B2, Annexin V+/PI+) through the damaged cell membranes. As shown in , there were group differences in the percentage of apoptotic cells among the groups (F (3, 12) = 16.341, P = 0.000). In line with the Nissl staining, the percentage of apoptotic cells (B2+B4) was significantly higher in the Pilo group than in the control group (P = 0.000), whereas the Pilo+KD group exhibited an obviously reduced apoptosis rate relative to the Pilo group (P = 0.030), suggesting that the KD protected the hippocampal cells from seizure-induced apoptosis. There was no significant difference in the apoptosis rates between the control group and KD alone group (P = 0.884).
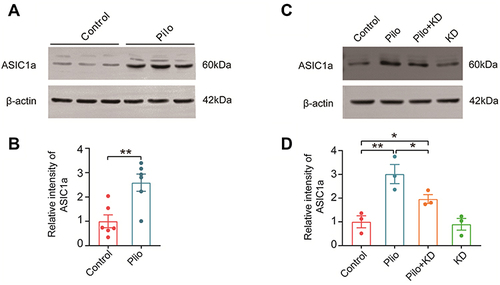
The KD Suppresses the Upregulation of ASIC1a in the Hippocampus of Rats with Chronic Seizures
Since Ca2+-permeable ASIC1a is largely responsible for acidosis-induced neuronal injury,Citation12 we first explored the expression of ASIC1a in the hippocampus of the chronic epileptic rats by Western blotting (). The results showed that the ASIC1a protein was obviously enhanced in the Pilo group compared with the control group (t (10) = 3.593, P = 0.005) (). The effect of the KD on the expression of ASIC1a was detected next (). Interestingly, significant differences in the expression of ASIC1a were observed among the four groups (F (3, 8) = 12.140, P = 0.002), and the upregulation of this protein was suppressed by the KD treatment to some extent (P = 0.030) (). In view of this, we speculated that ASIC1a may be involved in the neuroprotective effects of the KD.
Figure 5 The expression of ASIC1a protein in the hippocampus of rats with chronic seizures and the effect of the KD on it. (A) Representative Western blot images of ASIC1a in the control and Pilo groups. (B) Densitometric analysis of hippocampal ASIC1a expression in the control and Pilo groups (n=6 rats/group). (C) Representative Western blot images of ASIC1a in the control, Pilo, Pilo+KD and KD alone groups. (D) The densitometric analysis of the Western blot protein showed that recurrent seizures increased and that the KD suppressed the ASIC1a protein expression (n=3 rats/group). All values were normalised to the control samples and presented as mean ± SEM. *, **Represent P < 0.05 and P < 0.01, respectively. Abbreviations ASIC1a, acid-sensing ion channel 1a.
The KD Treatment Reduces Seizure-Related Intracellular Ca2+ Overload
Generally, Ca2+ entry through activated ASIC1a can disrupt the intracellular Ca2+ homeostasis, which contributes to neuronal injury.Citation32 Therefore, we monitored the [Ca2+]i in the hippocampal cells using Fluo-3 AM (). As illustrated by , significant differences in the [Ca2+]i among the groups were measured by flow cytometry (F (3, 12) = 17.584, P = 0.000). Moreover, a marked elevation of the [Ca2+]i was observed in the Pilo group compared with the control group (P = 0.000), while the seizure-induced intracellular Ca2+ overload was strikingly attenuated in the Pilo+KD group (P = 0.006). There was no significant difference in the [Ca2+]i between the control group and KD alone group (P = 0.934).
Figure 6 The [Ca2+]i, MMP, and mROS in the hippocampal cells in each group as assessed by flow cytometry assay. (A) The [Ca2+]i was detected by Fluo-3 AM. (B) The MMP was detected by JC-1. The shift of JC-1 fluorescence from aggregates (B2) to monomers (B4) indicated a collapse of the MMP. (C) The quantitative analysis of the [Ca2+]i was represented as the relative fluorescence intensity of Fluo-3 compared with the control group. (D) The quantitative analysis of MMP was represented as the percentage of JC-1 monomer-positive cells (B4). (E) The mROS were detected by DCFH-DA, and the quantitative analysis was represented as the relative fluorescence intensity of DCF compared with the control group. All results were expressed as the mean ± SEM (n = 4 rats/group). *, **, ***Represent P < 0.05, P < 0.01 and P < 0.001, respectively.
![Figure 6 The [Ca2+]i, MMP, and mROS in the hippocampal cells in each group as assessed by flow cytometry assay. (A) The [Ca2+]i was detected by Fluo-3 AM. (B) The MMP was detected by JC-1. The shift of JC-1 fluorescence from aggregates (B2) to monomers (B4) indicated a collapse of the MMP. (C) The quantitative analysis of the [Ca2+]i was represented as the relative fluorescence intensity of Fluo-3 compared with the control group. (D) The quantitative analysis of MMP was represented as the percentage of JC-1 monomer-positive cells (B4). (E) The mROS were detected by DCFH-DA, and the quantitative analysis was represented as the relative fluorescence intensity of DCF compared with the control group. All results were expressed as the mean ± SEM (n = 4 rats/group). *, **, ***Represent P < 0.05, P < 0.01 and P < 0.001, respectively.](/cms/asset/4fc35805-9364-4f74-9dd8-e49de7666c9c/dndt_a_12153215_f0006_c.jpg)
The KD Restores Mitochondrial Functions Following Seizures
Intracellular Ca2+ overload may result in loss of the MMP and accumulation of mROS, which are early events of seizure-induced apoptosis.Citation33 Thus, we investigated the potential role of the KD in MMP and mROS production in hippocampal cells using JC-1 staining and 2,7-dichlorofluorescein diacetate (DCFH-DA), respectively (). As illustrated by and , there were group differences in the levels of MMP (F (3, 12) = 21.153, P = 0.000) and mROS (F (3, 12) = 26.975, P = 0.000) among the four groups. Furthermore, the loss of MMP and release of mROS were dramatically increased in the Pilo group compared with the control group (MMP P = 0.000; mROS P = 0.000), while the increases were markedly inhibited by KD in the Pilo+KD group (MMP P = 0.002; mROS P = 0.006), demonstrating that the KD maintained the MMP and attenuated the mROS generation following the seizures. The KD alone group did not show significant differences in the MMP and mROS levels compared to the control group (MMP P = 0.952; mROS P = 0.743).
The KD Preserves the Neuronal Mitochondrial Ultrastructure Following Seizures
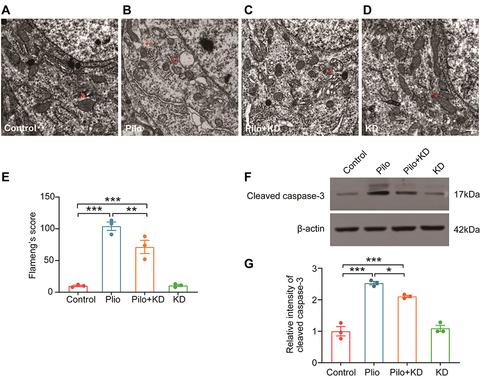
Further examination using a transmission electron microscope confirmed the protective effect of the KD on neuronal mitochondria (). The neuronal mitochondria of CA1 appeared normal, with intact mitochondrial membranes and cristae in both the control and KD alone groups (). In contrast, the mitochondria in the Pilo group were severely swollen and vacuolated, with rupturing cristae and membranes (). However, KD reduced the seizure induced injury of the mitochondria with only slight swelling and cristae separation (). In the quantitative analysis, significant differences (F (3, 8) = 55.127, P = 0.000) in the Flameng’s score of the mitochondria were observed among the four groups (). More specifically, the score in the Pilo group was obviously higher than that in the control group (104.00 ± 6.66 vs 10.00 ± 1.15, respectively; P = 0.000), while the mitochondria in the Pilo+KD group exhibited a lower score compared to those in the Pilo group (71.33 ± 10.48 vs 104.00 ± 6.66, respectively; P = 0.006).
Figure 7 Effects of the KD on the ultrastructure of the mitochondria and expression of cleaved caspase-3 following seizures. (A–D) The ultrastructure of the neuronal mitochondria in the CA1 area of the hippocampus was observed by transmission electron microscopy. Intact mitochondrial membranes and cristae could be observed in the mitochondria of the control (A) and KD alone (D) groups. The mitochondria in the Pilo group were severely swollen and vacuolated, with rupturing cristae and membranes (B); these abnormal changes were reversed by the KD treatment (C). Red arrows neuronal mitochondria. Scale bar = 500 nm. (E) Quantitative analysis of structurally damaged mitochondria with the Flameng’s score (n=3 rats/group). (F) Representative Western blot images of cleaved caspase-3 in each group. (G) Densitometric analysis of hippocampal cleaved caspase-3 expression in each group (n=3 rats/group). The data represented the relative quantity compared with the control group. All results were expressed as the mean ± SEM and analysed by a one-way ANOVA with the LSD test. *, **, ***Represent P < 0.05, P < 0.01 and P < 0.001, respectively.
The KD Inhibits the Overexpression of Cleaved Caspase-3 in the Hippocampus of Rats with Chronic Seizures
The expression of cleaved caspase-3, which is the effector caspase involved in mitochondria-related apoptosis, was measured by Western blotting (). As shown in , there were significant differences (F (3, 8) = 66.527, P = 0.000) in the level of cleaved caspase-3 among the groups. Moreover, it revealed that the expression of this protein in the hippocampus of the rats in the Pilo group increased markedly compared with that in the control group (P = 0.000), whereas the upregulation was suppressed by KD in the Pilo+KD group (P = 0.012), further confirming the inhibition of the mitochondrial apoptotic pathway.
Discussion
Epilepsy, which affects about 50 million people worldwide, is the third most common chronic brain disease. The genetics and aetiology of epilepsy are complex and there are a variety of potential genetic and epigenetic mechanisms.Citation34 TLE is one of the most common acquired epilepsy. Patients with epilepsy experience recurrent seizures due to abnormal neuronal activity in the brain. Given that TLE is generally refractory, it is important to understand the underlying mechanisms of epileptic development. Animal models that reproduce key features of human TLE provide a better understanding of the pathophysiology of TLE and allow us to easily monitor and manipulate the key factors that contribute to the development of epilepsy.Citation35 TLE animal models mainly include pilocarpine model, KA model, amygdala nuclear ignition model, 6 Hz corneal ignition model, and hereditary epilepsy animal model. However, each model has its own advantages and disadvantages compared with other models, and the appropriate model needs to be selected according to the experimental requirements.Citation36–40 All existing animal models have limitations, and they cannot fully simulate the characteristics of human TLE.
Chemical convulsant-induced SE has been widely used. Unlike other epilepsy models, systemic injection of chemical convulsant can simulate the clinical pathogenesis of human TLE, ie, initial brain injury, incubation period and chronic epileptic stage presenting with SRS. Therefore, this technique can be used for various studies to explain the mechanism of acute brain injury, occurrence of epilepsy, or inhibition of epilepsy. Furthermore, chemical convulsion-induced histopathological changes similar to those in human TLE provide additional justification for the use of TLE rodent models.Citation41
Our study showed that treatment with the KD for 28 days, starting after pilocarpine-induced SE, not only exerted antiepileptogenic effects but also exhibited excellent cognitive improvement and neuroprotective effects in the rats with TLE. Furthermore, the biochemical and flow cytometry results demonstrated that the KD inhibited seizure-induced upregulation of ASIC1a and the ensuing intracellular Ca2+ overload and restored the disrupted structure and function of the mitochondria. Collectively, these results indicated that ASIC1a and the mitochondrial pathway may play a crucial role in the neuroprotection mechanisms of the KD.
Epileptogenesis of TLE is often start from acute brain injuries, such as SE, hypoxia or traumatic brain injury, followed by a latent period, during which the brain experiences a cascade of morphological and functional alterations that eventually progress to SRSs.Citation42 Since the pilocarpine model reproduces the main characteristics of human TLE, it is regarded as a valuable tool to investigate the long-term efficacy of novel therapies for suppressing epileptogenesis as well as improving the cognitive and neuropathological changes associated with TLE.Citation23 The KD, a high-fat, low-carbohydrate and restricted-protein diet, is a well-known treatment for refractory epilepsy. Although the anti-seizure effects are understood to a certain extent, the antiepileptogenic properties of this diet must be further validated. Thus, we initiated KD treatment during the latent period following SE and found that the frequency and duration of the SRSs were attenuated after four weeks of treatment; however, the reduction of the duration was not obvious. Additionally, the spike amplitude of epileptic discharges was decreased to a certain extent after the KD. These findings were in agreement with those of previous studies, which demonstrated that the KD exerts significant antiepileptogenic effects in different seizure models.Citation43,Citation44
The recurrent seizures of TLE are often accompanied by cognitive co-morbidity, manifested as poor performance in several cognitive domains especially episodic memory, since the temporal lobe is the major structure involved in learning, memory and affective behaviour.Citation45,Citation46 Evidence indicates that the KD can improve cognitive function in both epilepsy patients and the pentylenetetrazol-induced epilepsy model.Citation4,Citation47 However, studies on the relationship between the KD and cognitive function in this model are scarce. Consistently, in this study, we observed that the rats exhibited severe spatial learning and memory impairments during the chronic period following SE. The KD improved the performance of epileptic rats in the MWM test, further confirming its effectiveness. This established a good foundation for the following research on the neuroprotective effects of this diet.
Seizure-related cognitive impairments are mostly associated with neurodegeneration and the selective loss of certain neuron populations, which are pathological characteristics of TLE.Citation8,Citation48 Furthermore, neurodegeneration and neuronal death are ongoing processes lasting throughout the entire duration of epilepsy, which means that cell death mechanisms are not only triggered by the occurrence of SE but also sustained by recurrent seizures.Citation49 This indicates that it is necessary to initiate neuroprotective treatment before the onset of SRSs. Here, from the results of the Nissl staining, we observed obvious loss and injury of the hippocampal neurons in epileptic rats on day 58 post-SE, and the CA1 region was more severe than the CA3 region. Moreover, counting the apoptotic cells by flow cytometry further confirmed the hippocampal injury induced by the seizures. This was consistent with previous research.Citation50,Citation51 However, KD effectively alleviated the neuronal degeneration and apoptosis following the seizures. Our results were sufficiently clear in indicating that the KD exhibited excellent neuroprotective effects in the chronic epileptic rats, though this may have been due to the reduction of the SRSs.
Acid-sensing ion channel 1a, which is Ca2+-permeable and highly sensitive to protons, has been demonstrated to contribute to acidosis-induced neurodegeneration.Citation12,Citation32 In TLE patients and epileptic mice, overexpressed ASIC1a in the reactive astrocytes of the hippocampus mediates Ca2+ influx, which facilitates the development of epileptogenesis.Citation17 However, ASIC1a can be activated not only in astrocytes but also in neurons when acidosis occurs in the same tissue. Moreover, ASIC1a-positive neurons are found to be swollen and highly vulnerable to seizure-induced cell death in epileptic rats and TLE patients, and inhibiting ASIC1a preserves the neuronal morphology and decreases neuronal loss.Citation19 In this study, high levels of ASIC1a were found in the hippocampus of the epileptic rats, proving the involvement of ASIC1a in the chronic period of TLE. Moreover, the overexpression of this protein was suppressed by the KD. This result coincided with the results of a previous experiment in vitro, which demonstrated that ketone bodies inhibit the activation of ASIC1a in rat hippocampal excitatory neurons.Citation22 The Ca2+ entry through ASIC1a is a prominent mechanism leading to neurodegeneration. In addition, ASIC1a also behaves like a gate controller to facilitate the activation of voltage-gated Ca2+ channels and NMDA receptor gated channels, which further promotes neuronal excitation and enhances ASIC1a-mediated intracellular Ca2+ accumulation.Citation20,Citation52,Citation53 Our data revealed elevations in the [Ca2+]i in the hippocampal cells of the epileptic rats, and the KD treatment attenuated the [Ca2+]i response. Therefore, the neuroprotective effects of the KD may occur via inhibiting the upregulation and activation of ASIC1a as well as the ensuing intracellular Ca2+ overload, which is a primary factor causing neuronal apoptosis.Citation8,Citation32,Citation54
Mitochondria are central to intracellular Ca2+ homeostasis, whereas excessively high [Ca2+]i can cause mitochondrial dysfunction, which plays a crucial role in the epileptogenesis and neuronal apoptosis associated with TLE.Citation55,Citation56 The intracellular Ca2+ overload induced by seizures causes continuous mitochondrial Ca2+ uptake, leading to MMP decrease, mROS generation and mitochondrial permeability transition pore (mPTP) opening, which could culminate in neuronal death.Citation33,Citation57 In addition, evidence indicates a crosstalk between ASIC1a and mitochondria under both physiological and pathophysiological conditions.Citation20,Citation21 In brief, in addition to acting as a proton-gated channel on the plasma membrane, ASIC1a is also expressed in mitochondria, mediates mitochondrial Ca2+ and Na+ influx leading to MMP depolarisation, and serves as an important regulator of mPTP opening.Citation20,Citation21,Citation58 In the current study, the KD showed clear mitochondrial protective effects, by alleviating the ultrastructural disruption of the neuronal mitochondria, restoring the MMP and preventing mROS accumulation in the epileptic rats. Together with the inhibition of caspase-3 activation, we demonstrated that the KD suppresses the mitochondria-dependent apoptotic pathway and that this effect may be associated with the inhibition of ASIC1a.
The neuroprotective effects induced by the KD may be one of the mechanisms underlying the antiepileptogenic properties of this diet. However, the exact role of ASIC1a in seizure generation, progression, and termination remains controversial. Multiple lines of evidence demonstrate that ASIC1a is highly expressed in excitatory neurons and reactive astrocytes, which contributes to seizure generation and epileptogenesis.Citation17,Citation22 Inhibiting ASIC1a with amiloride, a non-selective blocker, exhibits a remarkable anticonvulsant effect in pilocarpine, pentylenetetrazole, maximal electroshock and febrile seizure models.Citation59–62 Conversely, other studies have proposed that ASIC1a expression is decreased in the acute phase of epilepsyCitation19,Citation63 and that its activation is involved in seizure termination in kainate and pentylenetetrazole seizure models since acidosis may activate GABAergic interneurons through ASIC1a.Citation64 The seemingly contradictory role of ASIC1a in epilepsy may be associated with different seizure models and different periods of epilepsy. More specifically, the chemoconvulsant insult models (especially the pilocarpine model) induce serious damage to the hippocampus and progressive loss of GABAergic interneurons, which rarely occur in kindling models.Citation65,Citation66 Additionally, during the chronic period of epilepsy, reactive astrogliosis and significant loss of GABAergic interneurons predominate, which is rarely occur in the acute phase.Citation8,Citation67,Citation68 To elucidate the precise role of ASIC1a in epilepsy and determine whether the KD exerts antiepileptogenic effects through this channel, more knowledge on the integrated effects of ASIC1a in different neuronal cells is needed.
Conclusion
To recapitulate, our study provides a body of evidence demonstrating that the KD exerts not only antiepileptogenic effects but also excellent neuroprotective effects in rats with pilocarpine-induced TLE and that these effects likely occur via inhibiting ASIC1a and the downstream mitochondrial pathway. This extends the scope of current knowledge on the association between the KD and ASIC1a, and provides a theoretical basis for identifying novel therapeutic targets to halt hippocampal neurodegeneration and its related cognitive impairments in TLE. However, further studies are required to elaborate the specific molecular mechanisms underlying the effects of the KD on ASIC1a and mitochondria.
Ethics Approval
The experimental protocol was approved by the Animal Experimentation Ethics Committee of The Second Hospital of Hebei Medical University. Experimental animals underwent all procedures under anesthesia, and every effort was made to minimize their pain, suffering, and death.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
Acknowledgments
The authors wish to thank Academician Chunyan Li at the Key Laboratory of Neurology, the Second Hospital of Hebei Medical University for his invaluable assistance in providing the perfect experiment equipment and conditions.
Additional information
Funding
References
- Franco V, French JA, Perucca E. Challenges in the clinical development of new antiepileptic drugs. Pharmacol Res. 2016;103:95–104. doi:10.1016/j.phrs.2015.11.007
- Hermann BP, Seidenberg M, Dow C, et al. Cognitive prognosis in chronic temporal lobe epilepsy. Ann Neurol. 2006;60:80–87. doi:10.1002/ana.20872
- Lenck-Santini PP, Scott RC. Mechanisms responsible for cognitive impairment in epilepsy. Cold Spring Harb Perspect Med. 2015;5(10):a022772. doi:10.1101/cshperspect.a022772
- Wang X, Huang SP, Liu Y, et al. Effects of ketogenic diet on cognitive function in pentylenetetrazol-kindled rats. Epilepsy Res. 2021;170:106534. doi:10.1016/j.eplepsyres.2020.106534
- Murugan M, Boison D. Ketogenic diet, neuroprotection, and antiepileptogenesis. Epilepsy Res. 2020;167:106444. doi:10.1016/j.eplepsyres.2020.106444
- Noh HS, Kim YS, Choi WS. Neuroprotective effects of the ketogenic diet. Epilepsia. 2008;49(Suppl 8):120–123. doi:10.1111/j.1528-1167.2008.01855.x
- Blumcke I, Spreafico R, Haaker G, et al. Histopathological findings in brain tissue obtained during epilepsy surgery. N Engl J Med. 2017;377:1648–1656. doi:10.1056/NEJMoa1703784
- Walker MC. Hippocampal sclerosis causes and prevention. Semin Neurol. 2015;35:193–200. doi:10.1055/s-0035-1552618
- Gourmaud S, Shou HC, Irwin DJ, et al. Alzheimer-like amyloid and tau alterations associated with cognitive deficit in temporal lobe epilepsy. Brain. 2020;143:191–209. doi:10.1093/brain/awz381
- Alvarez de la Rosa D, Krueger SR, Kolar A, et al. Distribution, subcellular localization and ontogeny of ASIC1 in the mammalian central nervous system. J Physiol. 2003;546:77–87. doi:10.1113/jphysiol.2002.030692
- Alvarez de la Rosa D, Zhang P, Shao D, et al. Functional implications of the localization and activity of acid-sensitive channels in rat peripheral nervous system. Proc Natl Acad Sci U S A. 2002;992:326–331. doi:10.1073/pnas.042688199
- Xiong ZG, Zhu XM, Chu XP, et al. Neuroprotection in ischemia blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–698. doi:10.1016/j.cell.2004.08.026
- Xiong ZG, Pignataro G, Li MH, et al. Acid-sensing ion channels (ASICs) as pharmacological targets for neurodegenerative diseases. Curr Opin Pharmacol. 2008;8:25–32. doi:10.1016/j.coph.2007.09.001
- Siesjo BK, von Hanwehr R, Nergelius G, et al. Extra- and intracellular pH in the brain during seizures and in the recovery period following the arrest of seizure activity. J Cereb Blood Flow Metab. 1985;5:47–57. doi:10.1038/jcbfm.1985.7
- Somjen GG. Acidification of interstitial fluid in hippocampal formation caused by seizures and by spreading depression. Brain Res. 1984;311:186–188. doi:10.1016/0006-8993(84)91416-1
- Lv RJ, He JS, Fu YH, et al. ASIC1a polymorphism is associated with temporal lobe epilepsy. Epilepsy Res. 2011;96:74–80. doi:10.1016/j.eplepsyres.2011.05.002
- Yang F, Sun XL, Ding YX, et al. Astrocytic acid-sensing ion channel 1a contributes to the development of chronic epileptogenesis. Sci Rep. 2016;6:31581. doi:10.1038/srep31581
- Ievglevskyi O, Isaev D, Netsyk O, et al. Acid-sensing ion channels regulate spontaneous inhibitory activity in the hippocampus possible implications for epilepsy. Philos Trans R Soc Lond B Biol Sci. 2016;371(1700):20150431. doi:10.1098/rstb.2015.0431
- Wu H, Wang C, Liu B, et al. Altered expression pattern of acid-sensing ion channel isoforms in piriform cortex after seizures. Mol Neurobiol. 2016;53:1782–1793. doi:10.1007/s12035-015-9130-5
- Savic Azoulay I, Liu F, Hu Q, et al. ASIC1a channels regulate mitochondrial ion signaling and energy homeostasis in neurons. J Neurochem. 2020;153:203–215. doi:10.1111/jnc.14971
- Wang YZ, Zeng WZ, Xiao X, et al. Intracellular ASIC1a regulates mitochondrial permeability transition-dependent neuronal death. Cell Death Differ. 2013;201:359–369. doi:10.1038/cdd.2013.90
- Zhu F, Shan W, Xu QL, et al. Ketone bodies inhibit the opening of Acid-Sensing Ion Channels (ASICs) in rat hippocampal excitatory neurons in vitro. Front Neurol. 2019;10:155. doi:10.3389/fneur.2019.00155
- Curia G, Longo D, Biagini G, et al. The pilocarpine model of temporal lobe epilepsy. J Neurosci Methods. 2008;172:143–157. doi:10.1016/j.jneumeth.2008.04.019
- Smith ZZ, Benison AM, Bercum FM, et al. Progression of convulsive and nonconvulsive seizures during epileptogenesis after pilocarpine-induced status epilepticus. J Neurophysiol. 2018;119:1818–1835. doi:10.1152/jn.00721.2017
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi:10.1016/0013-4694(72)90177-0
- Su SW, Cilio MR, Sogawa Y, et al. Timing of ketogenic diet initiation in an experimental epilepsy model. Brain Res Dev Brain Res. 2000;125:131–138. doi:10.1016/s0165-3806(00)00130-9
- Sun C, Fu J, Qu ZZ, et al. Chronic mild hypoxia promotes hippocampal neurogenesis involving Notch1 signaling in epileptic rats. Brain Res. 2019;171:488–498. doi:10.1016/j.brainres.2019.02.011
- Sun C, Fu J, Qu ZZ, et al. Chronic intermittent hypobaric hypoxia restores hippocampus function and rescues cognitive impairments in chronic epileptic rats via wnt/beta-catenin signaling. Front Mol Neurosci. 2020;13:617143. doi:10.3389/fnmol.2020.617143
- Flameng W, Borgers M, Daenen W, et al. Ultrastructural and cytochemical correlates of myocardial protection by cardiac hypothermia in man. J Thorac Cardiovasc Surg. 1980;79:413–424.
- Krasemann S, Madore C, Cialic R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47(3):566–581.e9. doi:10.1016/j.immuni.2017.08.008
- Im EJ, Lee CH, Moon PG, et al. Sulfisoxazole inhibits the secretion of small extracellular vesicles by targeting the endothelin receptor A. Nat Commun. 2019;10(1):1387. doi:10.1038/s41467-019-09387-4
- Yermolaieva O, Soren Leonard A, Schnizler MK, et al. Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc Natl Acad Sci U S A. 2004;101:6752–6757. doi:10.1073/pnas.0308636100
- Kann O, Kovacs R. Mitochondria and neuronal activity. Am J Physiol Cell Physiol. 2007;292:C641–57. doi:10.1152/ajpcell.00222.2006
- Janson MT, Bainbridge JL. Continuing burden of refractory epilepsy. Ann Pharmacother. 2021;55(3):406–408. doi:10.1177/1060028020948056
- Ghannad-Rezaie M, Eimon PM, Wu Y, Yanik MF. Engineering brain activity patterns by neuromodulator polytherapy for treatment of disorders. Nat Commun. 2019;10(1):2620. doi:10.1038/s41467-019-10541-1
- Fu P, Yuan Q, Sun Y, et al. Baicalein ameliorates epilepsy symptoms in a pilocarpine-induced rat model by regulation of IGF1R. Neurochem Res. 2020;45(12):3021–3033. doi:10.1007/s11064-020-03150-8
- Mishra P, Mittal AK, Rajput SK, Sinha JK. Cognition and memory impairment attenuation via reduction of oxidative stress in acute and chronic mice models of epilepsy using antiepileptogenic Nux vomica. J Ethnopharmacol. 2021;267:113509. doi:10.1016/j.jep.2020.113509
- Mishra P, Sinha JK, Rajput SK. Efficacy of Cicuta virosa medicinal preparations against pentylenetetrazole-induced seizures. Epilepsy Behav. 2021;115:107653. doi:10.1016/j.yebeh.2020.107653
- Ghosh S, Sinha JK, Khan T, et al. Pharmacological and therapeutic approaches in the treatment of epilepsy. Biomedicines. 2021;9(5):470. doi:10.3390/biomedicines9050470
- Alese OO, Mabandla MV. Upregulation of hippocampal synaptophysin, GFAP and mGluR3 in a pilocarpine rat model of epilepsy with history of prolonged febrile seizure. J Chem Neuroanat. 2019;100:101659. doi:10.1016/j.jchemneu.2019.101659
- Singh T, Mishra A, Goel RK. PTZ kindling model for epileptogenesis, refractory epilepsy, and associated comorbidities: relevance and reliability. Metab Brain Dis. 2021;36(7):1573–1590. doi:10.1007/s11011-021-00823-3
- Pitkanen A, Lukasiuk K, Edward Dudek F, et al. Epileptogenesis. Cold Spring Harb Perspect Med. 2015;5(10):a022822. doi:10.1101/cshperspect.a022822
- Lusardi TA, Akula KK, Coffman SQ, et al. Ketogenic diet prevents epileptogenesis and disease progression in adult mice and rats. Neuropharmacology. 2015;99:500–509. doi:10.1016/j.neuropharm.2015.08.007
- Jiang Y, Yang Y, Wang S, et al. Ketogenic diet protects against epileptogenesis as well as neuronal loss in amygdaloid-kindling seizures. Neurosci Lett. 2012;508:22–26. doi:10.1016/j.neulet.2011.12.002
- Vrinda M, Arun S, Srikumar BN, et al. Temporal lobe epilepsy-induced neurodegeneration and cognitive deficits Implications for aging. J Chem Neuroanat. 2019;95:146–153. doi:10.1016/j.jchemneu.2018.02.005
- Bell B, Lin JJ, Seidenberg M, et al. The neurobiology of cognitive disorders in temporal lobe epilepsy. Nat Rev Neurol. 2011;7:154–164. doi:10.1038/nrneurol.2011.3
- Lambrechts DA, Wielders LHP, Aldenkamp AP, et al. The ketogenic diet as a treatment option in adults with chronic refractory epilepsy efficacy and tolerability in clinical practice. Epilepsy Behav. 2012;23:310–314. doi:10.1016/j.yebeh.2012.01.002
- Tai XY, Bernhardt B, Thom M, et al. Review Neurodegenerative processes in temporal lobe epilepsy with hippocampal sclerosis Clinical, pathological and neuroimaging evidence. Neuropathol Appl Neurobiol. 2018;44:70–90. doi:10.1111/nan.12458
- Nobili P, Colciaghi F, Finardi A, et al. Continuous neurodegeneration and death pathway activation in neurons and glia in an experimental model of severe chronic epilepsy. Neurobiol Dis. 2015;83:54–66. doi:10.1016/j.nbd.2015.08.002
- Luan G, Zhao YX, Zhai F, et al. Ketogenic diet reduces Smac/Diablo and cytochrome c release and attenuates neuronal death in a mouse model of limbic epilepsy. Brain Res Bull. 2012;89:79–85. doi:10.1016/j.brainresbull.2012.07.002
- Zhen JL, Wang WP, Zhou JJ, et al. Chronic intermittent hypoxic preconditioning suppresses pilocarpine-induced seizures and associated hippocampal neurodegeneration. Brain Res. 2014;1563:122–130. doi:10.1016/j.brainres.2014.03.032
- Gao S, Yu Y, Ma ZY, et al. NMDAR-mediated hippocampal neuronal death is exacerbated by activities of ASIC1a. Neurotox Res. 2015;281:22–37. doi:10.1007/s12640-015-9530-3
- Ma CL, Sun H, Yang L, et al. Acid-sensing ion channel 1a modulates NMDA receptor function through targeting NR1/NR2A/NR2B triheteromeric receptors. Neuroscience. 2019;406:389–404. doi:10.1016/j.neuroscience.2019.03.044
- Henshall DC. Apoptosis signalling pathways in seizure-induced neuronal death and epilepsy. Biochem Soc Trans. 2007;35:421–423. doi:10.1042/BST0350421
- Waldbaum S, Patel M. Mitochondria, oxidative stress, and temporal lobe epilepsy. Epilepsy Res. 2010;88:23–45. doi:10.1016/j.eplepsyres.2009.09.020
- Folbergrova J, Kunz WS. Mitochondrial dysfunction in epilepsy. Mitochondrion. 2012;12:35–40. doi:10.1016/j.mito.2011.04.004
- Rao VK, Carlson EA, Yan SS. Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochim Biophys Acta. 2014;1842:1267–1272. doi:10.1016/j.bbadis.2013.09.003
- Liu Z, Pei H, Zhang LM, et al. Mitochondria-targeted DNA nanoprobe for real-time imaging and simultaneous quantification of Ca(2+) and pH in neurons. ACS Nano. 2018;12:12357–12368. doi:10.1021/acsnano.8b06322
- Liang JJ, Huang LF, Chen XM, et al. Amiloride suppresses pilocarpine-induced seizures via ASICs other than NHE in rats. Int J Clin Exp Pathol. 2015;8:14507–14513.
- Ali A, Pillai KP, Ahmad FJ, et al. Anticonvulsant effect of amiloride in pentetrazole-induced status epilepticus in mice. Pharmacol Rep. 2006;58:242–245.
- Luszczki JJ, Sawicka KM, Kozinska J, et al. Amiloride enhances the anticonvulsant action of various antiepileptic drugs in the mouse maximal electroshock seizure model. J Neural Transm. 2009;116:57–66. doi:10.1007/s00702-008-0152-2
- Ou-Yang TP, Zhu GM, Ding YX, et al. The effects of amiloride on seizure activity, cognitive deficits and seizure-induced neurogenesis in a novel rat model of febrile seizures. Neurochem Res. 2016;41:933–942. doi:10.1007/s11064-015-1777-9
- Biagini G, Babinski K, Avoli M, et al. Regional and subunit-specific downregulation of acid-sensing ion channels in the pilocarpine model of epilepsy. Neurobiol Dis. 2001;8:45–58. doi:10.1006/nbdi.2000.0331
- Ziemann AE, Schnizler MK, Albert GW, et al. Seizure termination by acidosis depends on ASIC1a. Nat Neurosci. 2008;11:816–822. doi:10.1038/nn.2132
- Becker AJ. Review Animal models of acquired epilepsy insights into mechanisms of human epileptogenesis. Neuropathol Appl Neurobiol. 2018;44:112–129. doi:10.1111/nan.12451
- Covolan L, Mello LE. Temporal profile of neuronal injury following pilocarpine or kainic acid-induced status epilepticus. Epilepsy Res. 2000;39:133–152. doi:10.1016/s0920-1211(99)00119-9
- Wei D, Yang F, Wang Y, et al. Degeneration and regeneration of GABAergic interneurons in the dentate gyrus of adult mice in experimental models of epilepsy. CNS Neurosci Ther. 2015;21:52–60. doi:10.1111/cns.12330
- Magloczky Z, Freund TF. Impaired and repaired inhibitory circuits in the epileptic human hippocampus. Trends Neurosci. 2005;28:334–340. doi:10.1016/j.tins.2005.04.002