
Abstract
The 16p11.2 microdeletion syndrome is characterized by a wide range of phenotypic expressions and is frequently associated with developmental delay, symptoms from the autism spectrum, epilepsy, congenital anomalies, and obesity. These phenotypes are often related to a proximal 16p11.2 deletion of approximately 600 kb (BP4–BP5) that includes the SH2B1 gene that is reported to be causative for morbid obesity. This more centromeric deletion is most strongly related to autism spectrum susceptibility and is functionally different from the more distal 16p12.2p11.2 region, which includes the so-called atypical 16p11.2 BP2–BP3 deletion (approximately 220 kb) presenting with developmental delay, behavioral problems and mild facial dysmorphisms. Here, an adult male with a long history of maladaptive behaviors is described who was referred for diagnostic assessment of his amotivational features. Extensive neuropsychological examination demonstrated rigid thinking, anxious beliefs, and ideas of reference in the presence of normal intelligence. Microarray analysis demonstrated a de novo 970 kb 16p11.2 BP1–BP4 microdeletion that can be regarded as explanatory for his behavioral profile. It is concluded that microdeletion syndromes are not exclusively related to intellectual disabilities and genetic testing is of putative relevance for the understanding of neuropsychiatric and neuropsychological phenomena.
Introduction
Since high resolution comparative genomic hybridization techniques are increasingly available in the diagnostic procedures for patients with developmental disorders, several novel microdeletion and microduplication syndromes have been identified.Citation1–Citation3 One of these is the 16p11.2 microdeletion syndrome (Online Mendelian Inheritance in Man [OMIM]: 611913). As reviewed by the research groups of Battaglia and Barge-Schaapveld, the typical 16p11.2 microdeletion syndrome is characterized by a wide range of phenotypic expressions and is frequently associated with developmental delay, symptoms from the autism spectrum, epilepsy, congenital anomalies, and obesity.Citation4,Citation5 These phenotypes are often related to a proximal 16p11.2 deletion of approximately 600 kb (BP4–BP5) encompassing 29 genes,Citation6–Citation10 of which the SH2B1 gene (OMIM: 608937) is reported to be causative for morbid obesity.Citation11–Citation13 Battaglia et alCitation4 stated that this more centromeric deletion is most strongly related to autism spectrum susceptibility and is functionally different from the more distal region (16p12.2–p11.2; OMIM: 613604) which includes the so called atypical 16p11.2 deletion (approximately 220 kb) presenting with developmental delay, behavioral problems, and mild facial dysmorphisms.Citation5
In the present paper, an adult male with a long history of maladaptive behaviors, later associated with recurrent mild depressive and autistic symptoms, is described; the patient was referred for reevaluation of his clinical picture, particularly the amotivational features.
Patient and methods
Clinical description
The referred patient, a 33-year-old male, is the second son of non-consanguineous parents. He has a 5 years older, healthy brother. Apart from recurrent depression in his mother, there is no family history of intellectual disability, congenital anomalies, or neuropsychiatric disorders. The patient was born after an uncomplicated pregnancy and he had a normal birth weight and length (2,730 g and 48 cm, respectively; 0 standard deviation). No congenital anomalies were observed. There were normal developmental milestones. During early primary school years, both parents and teachers noticed social withdrawal tendencies and, incidentally, disproportionate reactions like clownish and impulsive behaviors to minor social stress, especially in novel and unstructured situations. Aged 16, a preliminary diagnosis of pervasive developmental disorder was made by a remedial educationalist at a community mental health center. Because of persistent behavioral problems, regularly accompanied by compelling and angry attitudes, secondary school education was fragmented; however, after several school changes, he finally completed high school at age 20. One year later, the patient discontinued his university studies because of problems with initiative and structuring of daily life. He was then admitted to a sheltered home facility. At the age of 24–25 years, marked motivational problems and recurrent mild depressive symptoms and/or ideas of reference necessitated several hospitalizations. Treatment with antidepressants and antipsychotics was given, without any effect. Ultimately, the patient was diagnosed with Asperger’s syndrome, a pervasive developmental disorder, and he was admitted to a specialized department for autism spectrum disorders. Neither somatic and neurological examination nor hematological and biochemical parameters disclosed any abnormalities. Electroencephalogram (EEG)-registration, magnetic resonance imaging (MRI), and dopamine transporter – single photon emission computed tomography (DAT-SPECT) of the brain were all normal. The diagnosis of Asperger’s disorder was confirmed and the patient subsequently entered a specialized sheltered home facility.
Present assessments
At the age of 32, he was referred for detailed reevaluation of his persistent amotivational behavior, for which no explanation could be given. Therefore, apart from somatic and neurological reexamination, extensive neuropsychological assessment including a Kaufman Adolescent and Adult Intelligence Test (KAIT) and a Minnesota Multiphasic Personality Inventory 2 Restructured Form (MMPI-2-RF) restructured clinical scale profiling was performed,Citation14,Citation15 as well as high density resolution, genome-wide array analysis.
Results
At examination, no dysmorphic features were found. Somatic and neurological examination did not disclose any abnormalities. Length and weight were 183 cm and 80 kg, respectively.
Neuropsychological profile
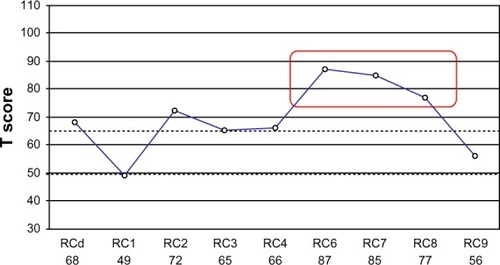
The patient’s behavior showed autistic-like elements such as diminished social reciprocity, paucity of speech, poor pragmatic language, and enhanced sensory perception. No major psychiatric symptoms could be detected. Neuropsychological assessment revealed a KAITCitation14 total intelligence quotient of 112 and undisturbed attention, memory, and executive functioning. MMPI-2-RF testingCitation15 disclosed poor reality testing, marked feelings of distrust, and an anxious disposition with excessive worrying (). This profile suggests an enhanced vulnerability to psychotic experiences.
Figure 1 MMPI-2-RF restructured clinical scale profile of the patient with a de novo, approximately 970 kb interstitial deletion in 16p11.2.
Abbreviations: MMPI-2-RF, Minnesota Multiphasic Personality Inventory 2 Restructured Form; RCd, Demoralization; RC1, Somatic Complaints; RC2, Low Positive Emotions; RC3, Cynicism; RC4, Antisocial Behavior; RC6, Persecutory Ideation; RC7, Dysfunctional Negative Emotions; RC8, Aberrant Experiences; RC9, Hypomanic Activation.
Affymetrix Cytoscan HD array
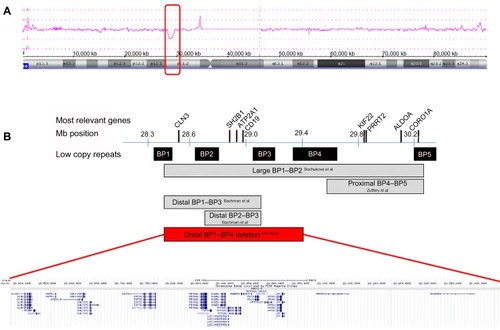
Genome wide array analysis with an average resolution of approximately 20 kb was performed using the Affymetrix CytoScan HD array platform (Affymetrix Inc., Santa Clara, CA, USA) with 2.6 million markers, including 750,000 genotype-able single nucleotide polymorphisms and 1.9 million nonpolymorphic probes. The array results showed a 970 kb microdeletion in the short arm of chromosome 16. Subsequent carrier testing by array analysis in the parents revealed that the 16p11.2 microdeletion had occurred de novo in their child, resulting in the karyotype: arr[hg19] 16p11.2(28,461,879-29,428,533)x1 dn. This novel 16p11.2 BP1–BP4 microdeletion encompasses 20 known protein coding genes and two microRNAs ().
Figure 2 Array plot of chromosome 16 and schematic representation of the distal 16p11.2 region.
Discussion
In the present paper, a 33-year-old, non-obese male with normal intelligence is described in whom a de novo, distal 16p11.2 BP1–BP4 microdeletion was demonstrated; this microdeletion can be regarded as causative for his pattern of anxiety-driven withdrawal behaviors, which result in recurrent episodes with referential thoughts. At this moment, no losses of similar size have been reported in healthy individuals (Database of Genomic Variants; http://dgv.tcag.ca/dgv/app/home).
So far, seven patients have been reported with a de novo microdeletion of 1.7–8.2 Mb in size, encompassing the 16p11.2 region and including the distal 16p11.2 deletion; in contrast, in six other patients a de novo distal (previously called atypical) 16p11.2 deletion was demonstrated (). Recently, Guha and et al reported the results of a large association study with a cohort of 790 patients with schizophrenia spectrum disorders.Citation16 They found 13 patients with 16p11.2 deletions ranging from BP1–BP4 which are not overlapping the proximal locus (BP4–BP5) that is connected to autism. This finding is in line with other copy-number variation studies and suggests this locus to be implicated in obesity and various neuropsychiatric disorders.Citation12,Citation17,Citation18
Table 1 Overview of patients with de novo 16p11.2 deletions (partially) overlapping the present deletion
The distal 16p11.2 deletion between BP1 and BP4, as reported here, encompasses 20 known protein-coding genes, ie, CLN3, APOBR, IL27, NUPR1, CCDC101, SULT1A2, SULT1A1, EIF3C, EIF3CL, ATXN2L, TUFM, SH2B1, ATP2A1, RABEP2, CD19, NFATC2IP, SPNS1, LAT, RRN3P2, and SNX29P2. Four of these are disease-causing genes: CD19 (OMIM: 107265); TUFM (OMIM: 602389); CLN3 (OMIM: 607042); and ATP2A1 (OMIM: 141750), without, however, any neuropsychiatric sequelae.
In conclusion, the distal 16p11.2 deletion phenotype is neither exclusively related to developmental delay nor to autism spectrum disorders. Rather, it has a highly variable presentation that may include, as demonstrated in the neuropsychological profile of the present patient, high average intelligence and social cognitive dysfunctions referring to Asperger’s syndrome.
Acknowledgments
The patient provided written informed consent for publication. This case has been submitted to the ECARUCA database (http://www.ecaruca.net) with identification number 5037. This study is part of a collaborative project of the research group “Psychopathology and Genetics” of the Radboud University Nijmegen and the Vincent van Gogh Institute for Psychiatry, Venray, the Netherlands.
Disclosure
The authors report no conflicts of interest in this work.
References
- VeltmanJAGenomic microarrays in clinical diagnosisCurr Opin Pediatr200618659860317099357
- SlavotinekAMNovel microdeletion syndromes detected by chromosome microarraysHum Genet2008124111718512078
- VissersLELMde LigtJGilissenCA de novo paradigm for mental retardationNat Genet201042121109111221076407
- BattagliaANovelliABernardiniLIgliozziRParriniBFurther characterization of the new microdeletion syndrome of 16p11.2–p12.2Am J Med Genet2009149A61200120419449418
- Barge-SchaapveldDQMaasSMPolstraAKnegtLCHennekamRCThe atypical 16p11.2 deletion: a not so atypical microdeletion syndrome?Am J Med Genet A2011155A51066107221465664
- WeissLAShenYKornJMAutism ConsortiumAssociation between microdeletion and microduplication at 16p11.2 and autismN Engl J Med2008358766767518184952
- SampsonMGCoughlinCRKaplanPEvidence for a recurrent microdeletion at chromosome 16p11.2 associated with congenital anomalies of the kidney and urinary tract (CAKUT) and Hirschsprung diseaseAm J Med Genet2010152A102618262220799338
- HansonENasirRHFongA16p11.2 Study Group CliniciansCognitive and behavioural characterization of 16p11.2 deletion syndromeJ Dev Behav Pediatr201031864965720613623
- CrepelASteyaertJDe la MarcheWNarrowing the critical deletion region for autism spectrum disorders on 16p11.2Am J Med Genet2011156224324521302354
- ZuffereyFSherrEHBeckmannNDSimons VIP Consortium; 16p11.2 European Consortium. A 600 kb deletion syndrome at 16p11.2 leads to energy imbalance and neuropsychiatric disordersJ Med Genet2012491066066823054248
- Bachmann-GageskuRMeffordHCCowanCRecurrent 200-kb deletions of 16p11.2 that include the SH2B1 gene are associated with developmental delay and obesityGenet Med2010121064164720808231
- BochukovaEGHuangNKeighJLarge, rare chromosomal deletions associated with severe early-onset obesityNature2010463728166667019966786
- WaltersRGJacquemontSValsesiaAA novel highly-penetrant form of obesity due to microdeletions on chromosome 16p11.2Nature2010463728167167520130649
- KaufmanSKaufmanNLMulderJLDekkerRDekkerPHDutch Adaptation of the Kaufman Adolescent and Adult Intelligence TestAmsterdamPearson2004
- van der HeijdenPTEggerJIDerksenJJPsychometric evaluation of the MMPI-2 Restructured Clinical scales in two Dutch samplesJ Pers Assess200890545646418704804
- GuhaSReesEDarvasiAMolecular Genetics of Schizophrenia Consortium; Wellcome Trust Case Control Consortium 2. Implication of a rare deletion at distal 16p11.2 in schizophreniaJAMA Psychiatry201370325326023325106
- CooperGMCoeBPGirirajanSA copy number variation morbidity map of developmental delayNat Genet201143983884621841781
- KirovGPocklingtonAJHolmansPDe novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophreniaMol Psychiatr2012172140153
- FirthHVRichardsSMBevanAPDECIPHER: Database of chromosomal imbalance and phenotype in humans using ensemble resourcesAm J Hum Genet200984452453319344873