
Abstract
Duchenne muscular dystrophy (DMD) is the most common form of muscular dystrophy in childhood. It is caused by mutations of the DMD gene, leading to progressive muscle weakness, loss of independent ambulation by early teens, and premature death due to cardiorespiratory complications. The diagnosis can usually be made after careful review of the history and examination of affected boys presenting with developmental delay, proximal weakness, and elevated serum creatine kinase, plus confirmation by muscle biopsy or genetic testing. Precise characterization of the DMD mutation is important for genetic counseling and individualized treatment. Current standard of care includes the use of corticosteroids to prolong ambulation and to delay the onset of secondary complications. Early use of cardioprotective agents, noninvasive positive pressure ventilation, and other supportive strategies has improved the life expectancy and health-related quality of life for many young adults with DMD. New emerging treatment includes viral-mediated microdystrophin gene replacement, exon skipping to restore the reading frame, and nonsense suppression therapy to allow translation and production of a modified dystrophin protein. Other potential therapeutic targets involve upregulation of compensatory proteins, reduction of the inflammatory cascade, and enhancement of muscle regeneration. So far, data from DMD clinical trials have shown limited success in delaying disease progression; unforeseen obstacles included immune response against the generated mini-dystrophin, inconsistent evidence of dystrophin production in muscle biopsies, and failure to demonstrate a significant improvement in the primary outcome measure, as defined by the 6-minute walk test in some studies. The long-term safety and efficacy of emerging treatments will depend on the selection of appropriate clinical end points and sensitive biomarkers to detect meaningful changes in disease progression. Correction of the underlying mutations using new gene-editing technologies and corticosteroid analogs with better safety profiles offers renewed hope for many individuals with DMD and their families.
Introduction – case illustration
A 3-year-old boy is referred for evaluation of developmental delay. He was born after an unremarkable pregnancy at term, with no complications. His development was normal during the first year. Although he is generally healthy and has good muscle bulk, he did not walk until after his second birthday. He is shy and he currently speaks in single words only. His examination is normal apart from calf hypertrophy and proximal weakness. He struggles to get up from the floor, and he is unable to run, jump, or climb stairs on his own. Initial investigation shows an elevated serum creatine kinase (CK) of 35,000 U/L (normal <200 U/L). Molecular genetic testing reveals an out of frame mutation in the DMD gene. His parents want to know more about the diagnosis and available treatment options.
Background on Duchenne muscular dystrophy
Duchenne muscular dystrophy (DMD) is a genetic muscle disorder that affects one per 3,500–5,000 live-born males; it is the most common type of muscular dystrophy in childhood.Citation1,Citation2 It is caused by mutations of the DMD gene, located on chromosome Xp21, which encodes for dystrophin, a 427 kDa protein that is expressed at the muscle sarcolemma. The absence of dystrophin destabilizes the muscle membrane, leading to the clinical features of motor developmental delay, calf hypertrophy, joint contractures, and progressive muscle weakness in affected boys, with markedly elevated serum CK that reflects ongoing muscle damage. In addition, boys with DMD may have a variable degree of speech delay, learning disability, and/or cognitive impairment.Citation3,Citation4 Progressive muscle degeneration eventually leads to loss of independent ambulation by early adolescence, scoliosis, cardiomyopathy, respiratory insufficiency, and reduced life expectancy, with death occurring before the third or fourth decade of life due to cardiorespiratory complications, according to recent DMD natural history studies.Citation5,Citation6
The DMD gene is one of the largest known human genes. It contains 79 exons, which include an actin-binding domain at the N-terminus, 24 spectrin-like repeat units, a cysteine-rich dystroglycan binding site, and a C-terminal domain.Citation7,Citation8 The extremely large size of the gene contributes to a complex mutational spectrum, with >7,000 different mutations and a high spontaneous mutation rate.Citation9 Approximately two-thirds of cases are maternally inherited; the remaining one-third occurs as a result of spontaneous mutations.Citation10 Large (one or more exons) deletions account for approximately two-thirds of all DMD mutations; the rest are due to duplications and small deletions, insertions, point mutations, or splicing mutations.Citation11,Citation12 The severe phenotype associated with DMD is most often caused by out-of-frame mutations, with complete loss of the dystrophin protein.Citation13,Citation14 In-frame mutations that allow for the synthesis of an internally truncated but partially functional protein are associated with a milder and more variable phenotype known as Becker muscular dystrophy or X-linked dilated cardiomyopathy; exceptions to the reading frame hypothesis occur in <10% of all DMD mutations.Citation10,Citation15 Disease severity is also affected by other genetic modifiers distinct from the DMD gene, including single nucleotide polymorphism of the latent transforming growth factor-beta (TGF-β) binding protein 4 (LBP4) gene and osteopontin, encoded by the secreted phosphoprotein 1 (SPP1) gene; both genes appear to influence disease progression in DMD by modifying the age at loss of independent ambulation, the age at onset of dilated cardiomyopathy, and the clinical response to corticosteroid treatment.Citation16–Citation18
Dystrophin is an integral part of the dystrophin-associated glycoprotein complex. It is in close association with other cytoskeletal proteins, including F-actin via its N-terminus and part of the rod domain; it also binds to dystroglycan via its cysteine-rich domain and to dystrobrevin and syntrophin via the C-terminal domain.Citation19 Thus, dystrophin provides structural stability to the skeletal muscle by connecting the sarcolemma and the basal lamina of the extracellular matrix to the inner cytoskeleton. It is also essential for cell survival via its transmembrane signaling function and modulation of vasomotor response to physical activity.Citation20 Three isoforms of dystrophin are derived from independent promoters in the brain, retina, and Purkinje cerebellar neurons; mutations in these tissue specific isoforms of dystrophin likely contribute to the extramuscular manifestations of DMD, including cognitive, behavioral, and learning difficulties.Citation19
Loss of dystrophin as a result of DMD gene mutations disrupts the dystrophin glycoprotein complex, leading to increased muscle membrane fragility. A cascade of events including influx of calcium into the sarcoplasm, activation of proteases and proinflammatory cytokines, and mitochondrial dysfunction results in progressive muscle degeneration.Citation20–Citation22 In addition, displacement of neuronal nitric oxide synthase contributes to tissue ischemia, increased oxidative stress, and reparative failure.Citation23 Disease progression is characterized by increasing muscle necrosis, fibrosis, and fatty tissue replacement and a greater degree of fiber size variation seen in subsequent muscle biopsies.Citation24
Making a diagnosis of DMD
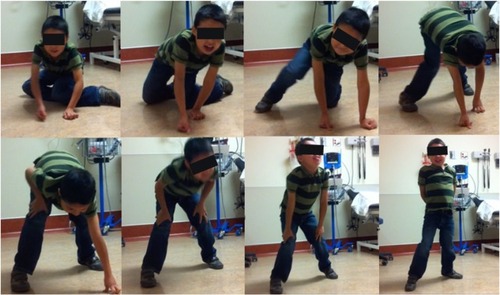
The diagnosis of DMD can usually be made after a careful review of the clinical history, physical examination, and confirmation by additional investigations, including muscle biopsy and/or molecular genetic testing.Citation3 A positive family history of DMD is not required, as approximately one-third of cases may occur as a result of spontaneous mutation. The presence of motor developmental delay with or without speech delay and muscle hypertrophy in a young boy should trigger the order of serum CK as an initial diagnostic screen for DMD, especially if the child also has signs of proximal muscle weakness, manifesting as an abnormal waddling gait, or a positive Gowers’ sign ().Citation25 As shown in the case illustration earlier, the muscle enzymes as measured by serum CK are usually markedly elevated. Raised muscle enzymes in DMD also contribute to persistently high serum alanine and aspartate transaminase levels; in some cases, the diagnosis may be delayed due to initial investigations for suspected hepatic dysfunction.
Figure 1 A young boy illustrates the typical Gowers’ sign.
Genetic testing for DMD mutations includes multiplex polymerase chain reaction focusing on the most commonly deleted regions of the gene or other assays that interrogate all 79 exons, such as multiplex ligation-dependent probe amplification (MLPA) or comparative genomic hybridization (CGH) microarray.Citation12,Citation26 If these diagnostic tests fail to identify the presence of a disease-causing deletion or duplication, complete gene sequencing is required to define the precise mutational event. A muscle biopsy can also be obtained for dystrophin immunostaining and extraction of RNA to produce complementary DNA and confirm a dystrophin mutation. The muscle biopsy typically shows a dystrophic process with ongoing muscle degeneration and regeneration; dystrophin immunostaining is usually absent or markedly reduced, except in rare revertant fibers. Using currently available diagnostic methods, it is possible to identify the mutations and confirm the clinical phenotypes in nearly all patients with dystrophinopathy.Citation27
Supportive treatment for DMD
Identification of a specific mutation is important for accurate diagnosis, prognosis, and individualized treatment for affected boys with DMD, as well as genetic counseling for their families.Citation28 Beyond realizing the severity of the diagnosis, parents live with uncertainty in anticipation of future losses and limitations that their children will experience, and anxiety as to how their children may react to those losses. For most parents, the demand of caring for their child with neuromuscular disease was disruptive to family life; the needs of the siblings, the spouse, and the primary caregiver tended to become secondary priorities.Citation29 The stress caused by the cognitive, behavioral, and other psychosocial challenges related to DMD can sometimes exceed those associated with the physical aspects of the disease.Citation30,Citation31 Increased burden of care and emotional distress are also common among caregivers of adults with DMD.Citation32 These realities underscore the need for assessment and support of the entire family.Citation3 Family-centered care, timely referrals, and appropriate community support can help parents to become experts on their child’s diagnosis and normalize their daily care routine ().Citation33 Mothers who are confirmed carriers of dystrophin mutations will also require periodic health surveillance for possible cardiomyopathy, myalgia, and/or proximal muscle weakness.Citation34
Table 1 Current supportive strategies for DMD
There is presently no cure for DMD. Current treatment strategies focus on optimizing growth and development, promoting well-balanced diet, participating in physical and recreational activity, and delaying the onset of secondary complications through ongoing medical and psychosocial support.Citation35 Supportive interventions including timely treatment with corticosteroids, early afterload reduction for cardiomyopathy, aggressive management of heart failure, noninvasive positive pressure ventilation, and effective airway clearance strategies have contributed to prolonged survival of individuals with DMD. The mean age of death from DMD increased from 14.4 years in the 1960s to 25.3 years in the 1990s, with corresponding improvement in affected individuals’ health-related quality of life.Citation36–Citation38
Standard of care for DMD
Recent publications from leading experts have provided comprehensive guidelines on the diagnosis and multidisciplinary management of DMD, including the use of corticosteroids.Citation3,Citation35,Citation39,Citation40 Historically, corticosteroids offered benefit to boys with DMD by stabilizing muscle strength and function,Citation41,Citation42 prolonging independent ambulation,Citation43,Citation44 and delaying the progression of scoliosisCitation45 and cardiomyopathy.Citation46 Continued treatment after loss of independent ambulation has also been shown to be beneficial.Citation47,Citation48 On the basis of available literature and clinical experience, current clinical practice guidelines strongly endorse the consideration of corticosteroid therapy for all DMD patients, starting at the early ambulatory stage of the disease.Citation3,Citation49 Daily oral prednisone (0.75 mg/kg) or deflazacort (0.9 mg/kg) is generally recommended as a disease-modifying treatment for DMD; intermittent steroids dosing schedule (10 days on/10 days off or high dose weekends only) are less commonly prescribed.Citation50 Prednisone and deflazacort are currently being studied in a head-to-head fashion as part of the FOR-DMD (NCT01603407) clinical trial; the results will hopefully determine the optimum corticosteroid regimen for DMD.
Common steroid-related side effects include short stature, obesity, cataracts, and skeletal fractures.Citation44,Citation51 In particular, approximately one-third of boys with DMD may develop vertebral compression fractures due to long-term corticosteroid use, progressive muscle weakness, impaired vitamin D and calcium absorption, and immobilization. Regular physical activity, calcium-enriched diet, optimization of vitamin D, and periodic assessments including lateral spine X-ray and bone density by dual energy X-ray absorptiometry scan are recommended as part of bone health management.Citation35 Bisphosphonates are generally reserved for those with symptomatic vertebral compression and/or recurrent fragility fractures in the extremity, in association with persistent or multiple bone health risk factors;Citation35 however, the long-term efficacy of bisphosphonate therapy for boys with DMD remains limited.Citation52,Citation53 Alternative treatments including denosumab and recombinant parathyroid hormone have not been formally evaluated in DMD.Citation54
Cardiac complications are common in DMD, including myocardial necrosis, conduction defects, and/or arrhythmias.Citation55,Citation56 Progressive cardiomyopathy is one of the leading causes of mortality in DMD.Citation36,Citation57 Common electro cardiography (ECG) abnormalities include sinus tachycardia, tall R waves in V1, deep Q waves in the inferolateral leads, ST depression, prolonged QT interval, and increased QT dispersion.Citation58 Echocardiographic evidence of structural heart disease in DMD patients includes left ventricular hypertrophy, regional wall motion abnormalities, dilation of the cardiac chambers, valvular abnormalities, and left ventricular systolic dysfunction.Citation59,Citation60 Despite the high prevalence of cardiac disease, most affected individuals are asymptomatic due to their low physical capability;Citation61 the diagnosis and treatment of DMD-related cardiomyopathy were often delayed.Citation62 According to the 2010 DMD guidelines, baseline assessment including ECG and echocardiogram should be performed at diagnosis, with reassessments at least every 2 years and then annually after 10 years of age.Citation35 However, echocardiograms may be technically difficult in DMD due to the development of progressive scoliosis, chest wall deformities, and respiratory insufficiency, thus limiting the utility of echocardiograms among patients with more advanced stages of the disease.
Increasingly, cardiac magnetic resonance imaging (MRI) has been used for the surveillance of myocardial dysfunction in DMD, especially in combination with gadolinium.Citation63 Using late gadolinium enhancement, cardiac MRI may detect early myocardial fibrosis, in the absence of any overt echocardiographic abnormalities;Citation64 however, young children (especially <6 years old) or those with significant cognitive or behavioral issues may not be able to tolerate the procedure without sedation. Early use of cardioprotective treatment including angiotensin-converting enzyme (ACE) inhibitors or beta-blocker may help delay the progression of cardiomyopathy.Citation35,Citation58 The addition of eplerenone to ACE inhibitors can also attenuate the decline in left ventricular systolic function.Citation65 According to a recent DMD working group, new cardiac guidelines will soon be available to address the expanded use of cardiac MRI, the use of myocardial strain analysis and other advanced echocardiographic imaging techniques, and the need for early initiation of anti-heart failure regimen for the treatment of DMD-related cardiomyopathy.Citation63
Individuals with DMD are also at increasing risk of respiratory complications due to progressive decline in respiratory muscle function.Citation66,Citation67 Signs of respiratory insufficiency include ineffective cough, recurrent chest infection, nocturnal hypoventilation, sleep disordered breathing, and eventually daytime respiratory failure.Citation36,Citation57 Regular pulmonary function testing is needed to monitor for early signs of respiratory insufficiency and to optimize treatment.Citation35 The initial treatment for nocturnal hypoventilation in DMD patients includes noninvasive positive pressure ventilation by nasal prongs or face mask to improve sleep quality, decrease daytime sleepiness, enhance gas exchange, and delay the decline in the pulmonary function.Citation68 Additional supportive strategies include chest physiotherapy, mechanical insufflation–exsufflation, and/or lung volume recruitment exercises to improve lung compliance and prevent atelectasis.Citation69,Citation70 The combination of noninvasive positive pressure ventilation, assisted airway clearance, mechanical cough assistance, and other respiratory supportive strategies allows DMD patients to live beyond their third decade of life.Citation71
The natural history of DMD includes increasing joint contractures and scoliosis as a result of progressive weakness, immobility, muscular imbalance, and fibrotic replacement in muscle tissue.Citation35 The use of corticosteroids is associated with prolonged independent ambulation, delayed onset of scoliosis, and reduced need for spinal surgery.Citation72 Affected individuals should be instructed to do active, active-assisted, and/or passive stretching exercise daily. Night splints can be worn to help minimize heel cord contractures, and serial casting may also be considered for short periods of time during the ambulatory phase of DMD.Citation35
Routine health surveillance and growth monitoring are important for all pediatric DMD patients. Optimal nutritional status, as defined by weight or body mass index between the 10th and the 85th percentiles for age, based on national growth charts, should be encouraged.Citation35 Referral to dietitian and formal feeding assessments may be indicated, particularly for those with bulbar symptoms or advanced stage of DMD.Citation73 Other anticipatory guidance includes regular trivalent inactivated influenza and 23-valent pneumococcal polysaccharide vaccinations, in addition to routine childhood immunizations. As well, assessment for wheelchair, lifts, and other adaptive technology should be included as part of the comprehensive treatment plan.Citation3,Citation35
Therapeutic strategies for DMD
Recent scientific advances have led to new disease-modifying treatments for many neuromuscular diseases including DMD.Citation74–Citation76 Identification of coordination centers and patients eligible for specific DMD trials has been greatly facilitated by the establishment of TREAT-NMD and other international DMD disease registries.Citation15 Regularly updated information about DMD clinical trials is available at https://www.ClinicalTrials.gov. A significant proportion of the active studies are enrolling participants by invitation only; as well, many of the studies listed are either Phase I safety or smaller Phase II pilot studies, with a fewer number of Phase III double-blind randomized trials. The main therapeutic strategies include, 1) gene replacement or other genetic therapies linked to specific mutations to restore dystrophin production;Citation77–Citation79 2) membrane stabilization and/or upregulation of compensatory proteins;Citation80,Citation81 and 3) reduction of the inflammatory cascade and/or enhancement of muscle regeneration ().Citation82,Citation83 Specific examples of emerging therapeutic strategies for DMD, including exon skipping and nonsense mutation suppression therapies, are briefly summarized in the genetic therapies section.
Table 2 Emerging therapies for DMD
Genetic therapies
Dystrophin gene replacement using virus vectors
Previous attempts to develop gene therapy for DMD have been complicated by the enormous size of the dystrophin gene. This was subsequently addressed by deleting selective regions of the dystrophin protein, leading to the generation of functional mini- and microdystrophins. Injection of adeno-associated viruses carrying microdystrophins into dystrophic canine model of DMD results in a striking improvement in the muscle histopathology.Citation84 However, earlier attempts in six boys with DMD due to frameshift mutations failed to achieve successful transgene expression, in part due to an unexpected primed T-cell-mediated immune response to the generated mini-dystrophin seen in two of the participants.Citation85 Additional clinical trials using recombinant adeno-associated viruses and more efficient vector delivery system to replace defective genes in DMD are currently in progress.Citation86
Exon skipping
Exon skipping uses synthetic antisense oligonucleotide sequences to correct specific dystrophin gene mutations. It does so by inducing specific exons skipping during pre-messenger RNA (pre-mRNA) splicing of the dystrophin gene, resulting in restoration of the reading frame and partial production of an internally truncated protein, similar to the dystrophin protein expression seen in Becker muscular dystrophy. Antisense therapies that induce single or multiple exon skipping could potentially be helpful for the majority of dystrophin mutations.Citation79
An earlier Phase I clinical trial of four boys with DMD using PRO051, a 2′-O-methyl-phosphorothioate oligoribonucleotide designed to skip exon 51 in dystrophin pre-mRNA, showed partial (17%–35%) restoration of dystrophin after a single intramuscular injection into the tibialis anterior.Citation87 This was followed by a Phase II study including weekly subcutaneous injections of PRO051, also known as drisapersen, up to 6 mg/kg/dose into 12 young DMD boys with a mean age of 9.2 years (range 5–13 years).Citation88 In addition to injection site reactions, drisapersen was associated with transient elevation of α1-microglubulinuria and variable proteinuria; a slight increase in dystrophin expression and a modest improvement in the mean 6-minute walk test (6MWT) distance of 35.2 m (SD 28.7 m) were seen after a 3-month open-label extension treatment.Citation88 Furthermore, a single subcutaneous injection of drisapersen at escalating doses for 20 nonambulatory boys aged 9–12 years with DMD was well tolerated, aside from transient proteinuria, fever, and acute inflammatory reactions seen in three participants given the 9 mg/kg dose.Citation89 However, a larger Phase III clinical trial involving 186 boys with DMD (NCT01254019) failed to show a significant difference in the 6MWT among participants on drisapersen versus placebo.Citation90 In a subsequent Phase II study involving a younger (mean age of 7.3 years, range 5–11 years) cohort of 53 patients with DMD, boys receiving continuous drisapersen weekly for 25 weeks demonstrated a slight improvement in the 6MWT and increased expression of dystrophin; however, there was no significant difference in the 6WMT at week 49.Citation91 Furthermore, results of formal muscle strength and other timed function tests did not differ between either continuous or intermittent drisapersen regimen versus placebo.Citation91 Further data from the open-label extension study of drisapersen (NCT02636686) is pending.
Eteplirsen (AVI-4658) is a phosphoramidate morpholino oligomer designed specifically for exon 51 skipping.Citation92,Citation93 In a subsequent double-blind placebo-controlled trial involving 12 participants (age 7–12 years) for 24 weeks followed by an open-label extension study of eteplirsen at either 30 mg/kg/wk or 50 mg/kg/wk, Mendell et alCitation94 found encouraging results in a subset (n=10) of participants, with significant improvement in walking ability and stabilization in their clinical function, and histological evidence of de novo dystrophin production and restoration of the dystrophin-associated protein complex in the muscle biopsies. Two older twin boys lost ambulation due to rapid disease progression, and they were excluded from the modified intention-to-treat analysis.Citation94 No significant adverse effects were noted after weekly treatment for >3 years following the open-label extension study.Citation95 The clinical benefits of eteplirsen for DMD will be determined as part of a larger confirmation study that is currently underway (PROMOVI, NCT0225552). Other exon skipping trials involving exons 44, 45, 53, and multiple exon skipping are planned.Citation96,Citation97
Nonsense suppression therapy
Approximately 10%–15% of DMD is caused by point mutations leading to a premature stop codon.Citation11,Citation15 Premature stop codons are nucleotide triplets within mRNA that signal the termination of protein translation by binding release factors and causing the ribosomal subunits to disassociate and release the shortened amino acid chain. This usually results in a loss of the functional protein, as critical parts of the amino acid chain are missing. Aminoglycosides and other nonsense mutation suppression agents bind to the ribosomal RNA subunits and impair the recognition of premature stop codon, thus allowing translation and production of a modified dystrophin protein. Earlier treatment trials with gentamicin showed mixed results, with additional caution regarding its long-term use due to potential renal- and oto-toxicity.Citation98–Citation100
Ataluren (also known as Translarna) is an orally bioavailable drug designed to overcome premature nonsense mutations.Citation101 Early studies of ataluren showed that it was generally safe and well tolerated.Citation102 Among 174 boys with DMD due to confirmed premature stop codon mutations, a double-blind placebo-controlled study for 48 weeks showed a marginally significant improvement in 6MWT for those receiving low-dose (40 mg/kg/d) ataluren only; those on high-dose (80 mg/kg/d) treatment did not improve in their 6MWT when compared with placebo, and the results were attributed to ataluren’s bell-shaped dose–response curve.Citation103 Ataluren has received conditional approval by the European Medicines Agency since August 2014;Citation104 results from the confirmatory trial (NCT02090959) is currently pending. Clinical trials involving other nonsense mutation suppression agents including arbekacin sulfate (NCT01918384) are being planned.Citation105
Cell therapy using muscle precursor cells or stem cells
Earlier attempts with myoblasts transfer were unsuccessful due to a number of factors, including limited viability of the donor cells after transplantation and suboptimal response from the use of older immunosuppressive regimen.Citation106 Even though a recent clinical trial showed that intra-arterial transplantation of human leukocyte antigen-matched sibling donor mesoangioblasts with better immunosuppression therapy was relatively safe and well tolerated, only a low level of donor DNA was found in the muscle biopsies, and no functional improvements were seen among the five boys with DMD posttransplant.Citation107 New cell therapies including induced pluripotent stem cells offer the potential of yielding an unlimited supply of autologous stem cells; however, patient-derived induced pluripotent stem cells will need to be genetically corrected before transplantation, and the long-term safety of this approach remains undefined.Citation108
Membrane stabilization and upregulation of cytoskeletal proteins
Compensatory upregulation of cytoskeleton proteins including utrophin,Citation109 alpha-7-beta-1 integrin,Citation110 biglycan,Citation111 and sarcospanCitation112 has been shown to stabilize the sarcolemma, in the absence of dystrophin in mdx mice, with improvement seen in the muscle biopsies. SMT C1100 is an oral bioavailable molecule specifically designed to target the utrophin-A promoter to increase utrophin expression; both SMT C1100 and its related compounds SMT022357 were shown in vitro and in vivo experiments to increase the production of utrophin and reduce the dystrophic changes in the skeletal and cardiac muscles.Citation113 Further clinical trials data related to SMT C1100 (NCT02056808 and NCT02383511) are currently pending.
Treatment of secondary cascades
Anti-inflammatory drugs
The IκB kinase/nuclear factor-kappa B (NF-κB) signaling is persistently elevated in immune cells and regenerative muscle fibers in both animal models and patients with DMD.Citation114 As well, activators of NF-κB such as tumor necrosis factor-α and interleukins 1 and 6 are upregulated in DMD muscles. Pharmacological inhibition of NF-κB using the NEMO-binding domain peptide resulted in improved pathology and muscle function in mouse models of muscular dystrophy.Citation115,Citation116 Additional research is needed to identify the role of selective NF-κB modulators and similar anti-inflammatory interventions for DMD.Citation117
Other potential anti-inflammatory therapies including N-acetylcysteine,Citation118 green tea extract,Citation119 idebenone,Citation120,Citation121 melatonin,Citation122,Citation123 and pentoxifyllineCitation124 were previously tried for DMD, with inconclusive results. Newer agents including givinostat (NCT01761292), a histone deacetylase inhibitor;Citation125 CAT1004 (NCT02439216), an oral molecule that inhibits activated NF-κB; and flavocoxid (NCT01335295), a blend of plant-derived flavonoids with anti-inflammatory activity, are currently under evaluation as potential disease-modifying treatment for DMD.Citation126,Citation127
Antifibrotic drugs
Elevated levels of TGF-β in muscular dystrophies stimulate fibrosis and impair muscle regeneration by blocking the activation of satellite cells. A number of antifibrotic agents have been tested in murine models of muscular dystrophy, including losartan, an angiotensin II-type 1 receptor blocker that reduces the expression of TGF-β.Citation128–Citation130 Other potential fibrosis inhibitors include HT-100 (halofuginone),Citation131 FG-3019, a monoclonal antibody to connective tissue growth factor,Citation132 and targeted microRNAs;Citation133,Citation134 further clinical trials are pending.
Muscle regeneration
Myostatin is a negative regulator of muscle mass. Inhibition or blockade of endogenous myostatin offers a potential means to compensate for the severe muscle wasting that is common in many types of muscular dystrophies including DMD. A Phase I/II multicenter clinical trial using MYO-029, a myostatin blocking antibody for adult subjects with Becker muscular dystrophies and other dystrophies, demonstrated safety, but the study was not sufficiently powered for efficacy.Citation74 Clinical trials using follistatin and other myostatin inhibitors including PF-06252616 (NCT02310764) and BMS-986089 (NCT02515669) are currently under way.Citation127,Citation135,Citation136 As well, the therapeutic potential of insulin growth factor (NCT01207908) as a positive regulator of muscle development and regeneration was shown in the dystrophic mouse models,Citation137 especially when combined with mesenchymal stromal cells.Citation138,Citation139 Due to its regeneration-enhancing mechanism, this combinational approach may have general applicability for other muscular dystrophies.
Treatment of muscle ischemia
Loss of dystrophin leads to displacement of neuronal nitric oxide synthase and reduction of muscle-derived nitric oxide to the microvasculature, resulting in functional muscle ischemia and further muscle injury.Citation19 Strategies to increase blood flow include pharmaceutical inhibition of either phosphodiesterase-5 or ACE, induction of angiogenesis through delivery of vascular endothelial growth factor (VEGF), or downregulation of the VEGF decoy-receptor type 1 (VEGFR-1 or Flt-1).Citation140,Citation141 Despite initial promising results,Citation142 clinical trials involving the use of tadalafil as disease-modifying treatment for DMD (NCT01865084 and 01070511) have recently been terminated due to a lack of demonstrated efficacy in slowing the decline of the 6MWT.
Future therapeutic targets
Given the limited success of exon skipping and other disease-modifying treatment for DMD to date, future clinical trials may include pharmacogenomics consideration, high-throughput screening, mRNA or genome-wide association studies, and additional translational research from animal models to identify other potential therapeutic options.Citation78,Citation143 Current challenges including T-cell-mediated immune response or antibodies to generated dystrophin, limited evidence of dystrophin production in muscle biopsies, inefficient methods of drug delivery to the whole body, and inconsistent efficacy will need to be resolved. The long-term safety and tolerability of ataluren, eteplirsen, and drisapersen will await additional monitoring and further Food and Drug Administration review. Currently, all three treatments have not been formally approved by the Food and Drug Administration due to insufficient data on long-term efficacy. Patient-focused perspective including health-related quality of life, satisfaction with care, acceptability or adherence to treatment, and participation in clinical trials will need to be considered in further collaborative research among neuromuscular disease registries and academic centers.
Correction of the genetic defect using engineered nucleases holds promise for the treatment of DMD.Citation144–Citation146 Recent studies on mdx mouse showed functional recovery in treated mice with RNA-guided clustered regularly interspaced short palindromic repeats-Cas9 endonucleases delivered by adeno-associated virus, with reversal of dystrophic changes in skeletal muscle fibers and cardiomyocytes, as well as in muscle satellite cells.Citation144,Citation147,Citation148 The same genome editing technology can potentially benefit the majority of individuals with DMD, especially when combined with newborn screening.Citation148 As well, a new clinical trial involving the study of VBP15, an oral glucocorticoid analog with anti-inflammatory properties and improved side-effect profile, is scheduled to begin later this year for ambulatory boys with DMD.Citation149
Conclusion
The pathogenesis affecting DMD is complex; multiple interventions targeting different disease processes are needed. Early recognition and precise genetic diagnosis will allow for individualized therapeutic options for DMD.Citation150 Even though there is presently no cure, respiratory intervention and other supportive strategies as outlined in the current standard of care for DMD have led to improved survival and better health-related quality of life for many affected individuals. New emerging treatments will depend on the appropriate use of clinical end points and sensitive surrogate outcome measures such as muscle MRI and circulating biomarkers to detect meaningful changes in disease progression.Citation151–Citation153
Disclosure
The author reports no conflicts of interest in this work.
References
- EmeryAEPopulation frequencies of inherited neuromuscular diseases – a world surveyNeuromuscul Disord19911119291822774
- MahJKKorngutLDykemanJDayLPringsheimTJetteNA systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophyNeuromuscul Disord201424648249124780148
- BushbyKFinkelRBirnkrantDJDiagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial managementLancet Neurol201091779319945913
- PaneMLombardoMEAlfieriPAttention deficit hyperactivity disorder and cognitive function in Duchenne muscular dystrophy: phenotype-genotype correlationJ Pediatr20121614705.e1709.e122560791
- McDonaldCMHenricsonEKAbreschRTThe cooperative international neuromuscular research group Duchenne natural history study – a longitudinal investigation in the era of glucocorticoid therapy: design of protocol and the methods usedMuscle Nerve2013481325423677550
- KienyPCholletSDelalandePEvolution of life expectancy of patients with Duchenne muscular dystrophy at AFM Yolaine de Kepper Centre between 1981 and 2011Ann Phys Rehabil Med201356644345423876223
- HoffmanEPBrownRHKunkelLMDystrophin: the protein product of the Duchenne muscular dystrophy locusCell19875169199283319190
- KoenigMMonacoAPKunkelLMThe complete sequence of dystrophin predicts a rod-shaped cytoskeletal proteinCell19885322192283282674
- BladenCLSalgadoDMongesSThe TREAT-NMD DMD Global Database: Analysis of More than 7,000 Duchenne Muscular Dystrophy MutationsHum Mutat201536439525604253
- Aartsma-RusAVan DeutekomJCFokkemaIFVan OmmenGJDen DunnenJTEntries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame ruleMuscle Nerve200634213514416770791
- MahJKSelbyKCampbellCA population-based study of dystrophin mutations in CanadaCan J Neurol Sci201138346547421515508
- NallamilliBRAnkalaAHegdeMMolecular diagnosis of Duchenne muscular dystrophyCurr Protoc Hum Genet2014839.25.19.25.2925271841
- KoenigMBeggsAHMoyerMThe molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletionAm J Hum Genet19894544985062491009
- MonacoAPBertelsonCJLiechti-GallatiSMoserHKunkelLMAn explanation for the phenotypic differences between patients bearing partial deletions of the DMD locusGenomics19882190953384440
- BladenCLRaffertyKStraubVThe TREAT-NMD Duchenne muscular dystrophy registries: conception, design, and utilization by industry and academiaHum Mutat201334111449145723913485
- PegoraroEHoffmanEPPivaLSPP1 genotype is a determinant of disease severity in Duchenne muscular dystrophyNeurology201176321922621178099
- BarpABelloLPolitanoLGenetic modifiers of Duchenne muscular dystrophy and dilated cardiomyopathyPLoS One20151010e014124026513582
- BelloLKesariAGordish-DressmanHGenetic modifiers of ambulation in the cooperative international neuromuscular research group Duchenne natural history studyAnn Neurol201577468469625641372
- MuntoniFTorelliSFerliniADystrophin and mutations: one gene, several proteins, multiple phenotypesLancet Neurol200321273174014636778
- WallaceGQMcNallyEMMechanisms of muscle degeneration, regeneration, and repair in the muscular dystrophiesAnnu Rev Physiol200971375718808326
- AllenDGGervasioOLYeungEWWhiteheadNPCalcium and the damage pathways in muscular dystrophyCan J Physiol Pharmacol2010882839120237582
- GumersonJDMicheleDEThe dystrophin-glycoprotein complex in the prevention of muscle damageJ Biomed Biotechnol2011201121079722007139
- ThomasGDFunctional muscle ischemia in Duchenne and Becker muscular dystrophyFront Physiol2013438124391598
- ShinJTajrishiMMOguraYKumarAWasting mechanisms in muscular dystrophyInt J Biochem Cell Biol201345102266227923669245
- van RuitenHJStraubVBushbyKGuglieriMImproving recognition of Duchenne muscular dystrophy: a retrospective case note reviewArch Dis Child201499121074107725187493
- HedgeMRChinELHMulleJGOkouDTWarrenSTZwickMEMicroarray-based mutation detection in the dystrophin geneHum Mutat20082991091109918663755
- TakeshimaYYagiMOkizukaYMutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral centerJ Hum Genet201055637938820485447
- Aartsma-RusAGinjaarIBBushbyKThe importance of genetic diagnosis for Duchenne muscular dystrophyJ Med Genet201653314515126754139
- MahJKThannhauserJEMacNeilDADeweyDBeing the lifeline: the parent experience of caring for a child with neuromuscular disease on home mechanical ventilationNeuromuscul Disord2008181298398818974004
- NereoNEFeeRJHintonVJParental stress in mothers of boys with Duchenne muscular dystrophyJ Pediatr Psychol200328747348412968039
- SamsonATomiakEDimilloJThe lived experience of hope among parents of a child with Duchenne muscular dystrophy: perceiving the human being beyond the illnessChronic Illn20095210311419474233
- PangalilaRFvan den BosGAStamHJvan ExelNJBrouwerWBRoebroeckMESubjective caregiver burden of parents of adults with Duchenne muscular dystrophyDisabil Rehabil2012341298899622149389
- McMillanHJCampbellCMahJKCanadian Paediatric Neuromuscular GroupDuchenne muscular dystrophy: Canadian paediatric neuromuscular physicians surveyCan J Neurol Sci201037219520520437929
- SoltanzadehPFriezMJDunnDClinical and genetic characterization of manifesting carriers of DMD mutationsNeuromuscul Disord201020849950420630757
- BushbyKFinkelRBirnkrantDJDiagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary careLancet Neurol20109217718919945914
- EagleMBaudouinSVChandlerCGiddingsDRBullockRBushbyKSurvival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilationNeuromuscul Disord2002121092692912467747
- Center for Disease ControlSurvival of males diagnosed with Duchenne/Becker muscular dystrophy (DBMD) by years of birth – muscular dystrophy surveillance tracking and research networkMMWR200958401119112219834452
- VillanovaMBrancalionBMehtaADDuchenne muscular dystrophy: life prolongation by noninvasive ventilatory supportAm J Phys Med Rehabil201493759559924743468
- BushbyKMuntoniFUrtizbereaAHughesRGriggsRReport on the 124th ENMC international workshop. Treatment of Duchenne muscular dystrophy; defining the gold standards of management in the use of corticosteroids. 2–4 April 2004, Naarden, the NetherlandsNeuromuscul Disord2004148–952653415336694
- MoxleyRTAshwalSPandyaSQuality Standards Subcommittee of the American Academy of NeurologyPractice Committee of the Child Neurology SocietyPractice parameter: corticosteroid treatment of Duchenne dystrophy: report of the quality standards subcommittee of the American Academy of Neurology and the practice committee of the Child Neurology SocietyNeurology2005641132015642897
- MendellJRMoxleyRTGriggsRCRandomized, double-blind six-month trial of prednisone in Duchenne’s muscular dystrophyN Engl J Med198932024159215972657428
- GriggsRCMoxleyRTMendellJRPrednisone in Duchenne dystrophy. A randomized, controlled trial defining the time course and dose response. Clinical investigation of Duchenne dystrophy groupArch Neurol19914843833882012511
- BiggarWDGingrasMFehlingsDLHarrisVASteeleCADeflazacort treatment of Duchenne muscular dystrophyJ Pediatr20011381455011148511
- ScharaUMortierJMortierWLong-term steroid therapy in Duchenne muscular dystrophy-positive results versus side effectsJ Clin Neuromuscul Dis20012417918319078632
- KinaliMMainMEliahooJPredictive factors for the development of scoliosis in Duchenne muscular dystrophyEur J Paediatr Neurol200711316016617257866
- MarkhamLWKinnettKWongBLWoodrow BensonDCripeLHCorticosteroid treatment retards development of ventricular dysfunction in Duchenne muscular dystrophyNeuromuscul Disord200818536537018436445
- HenricsonEKAbreschRTCnaanAThe Cooperative International Neuromuscular Research Group Duchenne natural history study: glucocorticoid treatment preserves clinically meaningful functional milestones and reduces rate of disease progression as measured by manual muscle testing and other commonly used clinical trial outcome measuresMuscle Nerve2013481556723649481
- PaneMFanelliLMazzoneESBenefits of glucocorticoids in non-ambulant boys/men with Duchenne muscular dystrophy: a multicentric longitudinal study using the performance of upper limb testNeuromuscul Disord2015251074975326248957
- GlossDMoxleyRTAshwalSOskouiMPractice guideline update summary: corticosteroid treatment of Duchenne muscular dystrophy: report of the guideline development subcommittee of the American Academy of NeurologyNeurology201686546547226833937
- AngeliniCPeterleEOld and new therapeutic developments in steroid treatment in Duchenne muscular dystrophyActa Myol201231191522655511
- McAdamLCMayoALAlmanBABiggarWDThe Canadian experience with long-term deflazacort treatment in Duchenne muscular dystrophyActa Myol2012311162022655512
- SbrocchiAMRauchFJacobPThe use of intravenous bisphosphonate therapy to treat vertebral fractures due to osteoporosis among boys with Duchenne muscular dystrophyOsteoporos Int201223112703271122297733
- HoustonCMathewsKShibli-RahhalABone density and alendronate effects in Duchenne muscular dystrophy patientsMuscle Nerve201449450651123835890
- BucknerJLBowdenSAMahanJDOptimizing bone health in Duchenne muscular dystrophyInt J Endocrinol2015201592838526124831
- American Academy of Pediatrics Section on Cardiology and Cardiac SurgeryCardiovascular health supervision for individuals affected by Duchenne or Becker muscular dystrophyPediatrics200511661569157316322188
- JudgeDPKassDAThompsonWRWagnerKRPathophysiology and therapy of cardiac dysfunction in Duchenne muscular dystrophyAm J Cardiovasc Drugs201111528729421812510
- PassamanoLTagliaAPalladinoAImprovement of survival in Duchenne muscular dystrophy: retrospective analysis of 835 patientsActa Myol201231212112523097603
- SpurneyCFCardiomyopathy of Duchenne muscular dystrophy: current understanding and future directionsMuscle Nerve201144181921674516
- BilchickKCSalernoMPlittDPrevalence and distribution of regional scar in dysfunctional myocardial segments in Duchenne muscular dystrophyJ Cardiovasc Magn Reson20111312021396105
- RomfhAMcNallyEMCardiac assessment in Duchenne and Becker muscular dystrophiesCurr Heart Fail Rep20107421221820857240
- NigroGComiLIPolitanoLBainRJThe incidence and evolution of cardiomyopathy in Duchenne muscular dystrophyInt J Cardiol19902632712772312196
- SpurneyCShimizuRMorgenrothLPCooperative international neuromuscular research group Duchenne natural history study demonstrates insufficient diagnosis and treatment of cardiomyopathy in Duchenne muscular dystrophyMuscle Nerve201450225025624395289
- McNallyEMKaltmanJRBensonDWContemporary cardiac issues in Duchenne muscular dystrophy. Working group of the national heart, lung, and blood institute in collaboration with parent project muscular dystrophyCirculation2015131181590159825940966
- HorKNTaylorMDAl-KhalidiHRPrevalence and distribution of late gadolinium enhancement in a large population of patients with Duchenne muscular dystrophy: effect of age and left ventricular systolic functionJ Cardiovasc Magn Reson20131510724359596
- RamanSVHorKNMazurWEplerenone for early cardiomyopathy in Duchenne muscular dystrophy: a randomised, double-blind, placebo-controlled trialLancet Neurol201514215316125554404
- FinderJDBirnkrantDCarlJRespiratory care of the patient with Duchenne muscular dystrophy: ATS consensus statementAm J Respir Crit Care Med2004170445646515302625
- BirnkrantDJBushbyKMAminRSThe respiratory management of patients with Duchenne muscular dystrophy: a DMD care considerations working group specialty articlePediatr Pulmonol201045873974820597083
- BachJRMartinezDDuchenne muscular dystrophy: continuous noninvasive ventilatory support prolongs survivalRespir Care201156674475021333078
- McKimDAKatzSLBarrowmanNNiALeBlancCLung volume recruitment slows pulmonary function decline in Duchenne muscular dystrophyArch Phys Med Rehabil20129371117112222421625
- BachJRSinqueeDMSaporitoLRBotticelloALEfficacy of mechanical insufflation-exsufflation in extubating unweanable subjects with restrictive pulmonary disordersRespir Care201560447748325492956
- LoMauroAD’AngeloMGAlivertiAAssessment and management of respiratory function in patients with Duchenne muscular dystrophy: current and emerging optionsTher Clin Risk Manag2015111475148826451113
- HsuJDQuinlivanRScoliosis in Duchenne muscular dystrophy (DMD)Neuromuscul Disord201323861161723746543
- DavidsonZETrubyHA review of nutrition in Duchenne muscular dystrophyJ Hum Nutr Diet200922538339319743977
- WagnerKRApproaching a new age in DMD treatmentNeurotherapeutics20085458359119019310
- MercuriEMuntoniFMuscular dystrophy: new challenges and review of the current clinical trialsCurr Opin Pediatr201325670170724240289
- ShiehPBDuchenne muscular dystrophy: clinical trials and emerging tribulationsCurr Opin Neurol201528554254626280938
- KoniecznyPSwiderskiKChamberlainJSGene and cell-mediated therapies for muscular dystrophyMuscle Nerve201347564966323553671
- BertoniCEmerging gene editing strategies for Duchenne muscular dystrophy targeting stem cellsFront Physiol2014514824795643
- Al-ZaidySRodino-KlapacLMendellJRGene therapy for muscular dystrophy: moving the field forwardPediatr Neurol201451560761825439576
- MalikVRodino-KlapacLRMendellJREmerging drugs for Duchenne muscular dystrophyExpert Opin Emerg Drugs201217226127722632414
- MarshallJLCrosbie-WatsonRHSarcospan: a small protein with large potential for Duchenne muscular dystrophySkelet Muscle201331123282144
- De PaepeBDe BleeckerJLCytokines and chemokines as regulators of skeletal muscle inflammation: presenting the case of Duchenne muscular dystrophyMediators Inflamm201320132–354037024302815
- MotohashiNAsakuraAMuscle satellite cell heterogeneity and self-renewalFront Cell Dev Biol20142125364710
- ShinJHPanXHakimCHMicrodystrophin ameliorates muscular dystrophy in the canine model of duchenne muscular dystrophyMol Ther201321475075723319056
- MendellJRCampbellKRodino-KlapacLDystrophin immunity in Duchenne’s muscular dystrophyN Engl J Med2010363151429143720925545
- OkadaTTakedaSCurrent challenges and future directions in recombinant aav-mediated gene therapy of Duchenne muscular dystrophyPharmaceuticals (Basel)20136781383624276316
- van DeutekomJCJansonAAGinjaarIBLocal dystrophin restoration with antisense oligonucleotide PRO051N Engl J Med2007357262677268618160687
- GoemansNMTuliniusMvan den AkkerJTSystemic administration of PRO051 in Duchenne’s muscular dystrophyN Engl J Med2011364161513152221428760
- FlaniganKMVoitTRosalesXQPharmacokinetics and safety of single doses of drisapersen in non-ambulant subjects with Duchenne muscular dystrophy: results of a double-blind randomized clinical trialNeuromuscul Disord2014241162424321374
- GoemansNCampbellCKrausJEDrisapersen efficacy and safety in Duchenne muscular dystrophy: results of a Phase III, randomized, double-blind, placebo-controlled trial (study DMD114044)World Muscle Society Congress [abstract]Asilomar, CA, USAOctober 1–5, 2013
- VoitTTopalogluHStraubVSafety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): an exploratory, randomised, placebo-controlled phase 2 studyLancet Neurol2014131098799625209738
- KinaliMArechavala-GomezaVFengLLocal restoration of dystrophin expression with the morpholino oligomer AVI-4658 in duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept studyLancet Neurol200981091892819713152
- CirakSArechavala-GomezaVGuglieriMExon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation studyLancet2011378979159560521784508
- MendellJRRodino-KlapacLRSahenkZEteplirsen for the treatment of Duchenne muscular dystrophyAnn Neurol201374563764723907995
- MendellJRGoemansNLowesLPLongitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophyAnn Neurol201679225727126573217
- MaluekaRGDwianingsihEKYagiMPhosphorothioate modification of chimeric 2′-o-methyl RNA/ethylene-bridged nucleic acid oligonucleotides increases dystrophin exon 45 skipping capability and reduces cytotoxicityKobe J Med Sci2015604E86E9425791417
- YuXBaoBEchigoyaYYokotaTDystrophin-deficient large animal models: translational research and exon skippingAm J Transl Res2015781314133126396664
- WagnerKRHamedSHadleyDWGentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutationsAnn Neurol200149670671111409421
- PolitanoLNigroGNigroVGentamicin administration in Duchenne patients with premature stop codon. Preliminary resultsActa Myol2003221152112966700
- MalikVRodino-KlapacLRViolletLGentamicin-induced readthrough of stop codons in Duchenne muscular dystrophyAnn Neurol201067677178020517938
- WelchEMBartonERZhuoJPTC124 targets genetic disorders caused by nonsense mutationsNature20074477140879117450125
- FinkelRSFlaniganKMWongBPhase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dystrophyPLoS One2013812e8130224349052
- BushbyKFinkelRWongBAtaluren treatment of patients with nonsense mutation dystrophinopathyMuscle Nerve201450447748725042182
- HaasMVlcekVBalabanovPEuropean medicines agency review of Ataluren for the treatment of ambulant patients aged 5 years and older with Duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin geneNeuromuscul Disord201525151325497400
- KarijolichJYuYTTherapeutic suppression of premature termination codons: mechanisms and clinical considerations (review)Int J Mol Med201434235536224939317
- AsakuraASkeletal muscle-derived hematopoietic stem cells: muscular dystrophy therapy by bone marrow transplantationJ Stem Cell Res Ther2012Suppl 1115
- CossuGPrevitaliSCNapolitanoSIntra-arterial transplantation of hla-matched donor mesoangioblasts in Duchenne muscular dystrophyEMBO Mol Med20157121513152826543057
- BriggsDMorganJERecent progress in satellite cell/myoblast engraftment – relevance for therapyFEBS J2013280174281429323560812
- TinsleyJMFaircloughRJStorerRDaily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mousePLoS One201165e1918921573153
- HellerKNMontgomeryCLJanssenPMClarkKRMendellJRRodino-KlapacLRAAV-mediated overexpression of human α7 integrin leads to histological and functional improvement in dystrophic miceMol Ther201321352052523319059
- AmentaARYilmazABogdanovichSBiglycan recruits utrophin to the sarcolemma and counters dystrophic pathology in mdx miceProc Natl Acad Sci U S A2011108276276721187385
- MarshallJLKwokYMcMorranBJBaumLGCrosbie-WatsonRHThe potential of sarcospan in adhesion complex replacement therapeutics for the treatment of muscular dystrophyFEBS J2013280174210422923601082
- GuiraudSSquireSEEdwardsBSecond-generation compound for the modulation of utrophin in the therapy of DMDHum Mol Genet201524154212422425935002
- AcharyyaSVillaltaSABakkarNInterplay of IKK/NF-KappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophyJ Clin Invest2007117488990117380205
- PetersonJMKlineWCananBDPeptide-based inhibition of NF-κB rescues diaphragm muscle contractile dysfunction in a murine model of Duchenne muscular dystrophyMol Med2011175–650851521267511
- DelfínDAXuYPetersonJMGuttridgeDCRafael-FortneyJAJanssenPMImprovement of cardiac contractile function by peptide-based inhibition of NF-κB in the utrophin/dystrophin-deficient murine model of muscular dystrophyJ Transl Med201196821586145
- UrsoMLAnti-inflammatory interventions and skeletal muscle injury: benefit or detriment?J Appl Physiol2013115692092823539314
- WhiteheadNPPhamCGervasioOLAllenDGN-Acetylcysteine ameliorates skeletal muscle pathophysiology in mdx miceJ Physiol200858672003201418258657
- EvansNPCallJABassaganya-RieraJRobertsonJLGrangeRWGreen tea extract decreases muscle pathology and NF-kappa B immunostaining in regenerating muscle fibers of mdx miceClin Nutr201029339139819897286
- BuyseGMVan der MierenGErbMLong-term blinded placebo-controlled study of SNT-MC17/idebenone in the dystrophin deficient mdx mouse: cardiac protection and improved exercise performanceEur Heart J200930111612418784063
- BuyseGMGoemansNvan den HauweMIdebenone as a novel, therapeutic approach for Duchenne muscular dystrophy: results from a 12 month, double-blind, randomized placebo-controlled trialNeuromuscul Disord201121639640521435876
- HibaouiYReutenauer-PatteJPatthey-VuadensORueggUTDorchiesOMMelatonin improves muscle function of the dystrophic mdx5cv mouse, a model for Duchenne muscular dystrophyJ Pineal Res201151216317121486366
- ChahbouniMEscamesGLópezLCMelatonin treatment counteracts the hyperoxidative status in erythrocytes of patients suffering from Duchenne muscular dystrophyClin Biochem20114410–1185385821515247
- EscolarDMZimmermanABertoriniTPentoxifylline as a rescue treatment for DMD: a randomized double-blind clinical trialNeurology2012781290491322402864
- ConsalviSMozzettaCBetticaPPreclinical studies in the mdx mouse model of Duchenne muscular dystrophy with the histone deacetylase inhibitor givinostatMol Med201319798723552722
- WyattEJSweeneyHLMcNallyEMMeeting report: new directions in the biology and disease of skeletal muscle 2014J Neuromuscul Dis20141219720626207203
- HeslopECsimmaCStraubVThe TREAT-NMD advisory committee for therapeutics (TACT): an innovative de-risking model to foster orphan drug developmentOrphanet J Rare Dis2015104925902795
- CohnRDvan ErpCHabashiJPAngiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic statesNat Med200713220421017237794
- SpurneyCFSaliAGuerronADLosartan decreases cardiac muscle fibrosis and improves cardiac function in dystrophin-deficient mdx miceJ Cardiovasc Pharmacol Ther2011161879521304057
- BishLTYarchoanMSleeperMMChronic losartan administration reduces mortality and preserves cardiac but not skeletal muscle function in dystrophic micePLoS One201166e2085621731628
- BodanovskyAGuttmanNBarzilai-TutschHHalofuginone improves muscle-cell survival in muscular dystrophiesBiochim Biophys Acta2014184371339134724703880
- KharrazYGuerraJPessinaPSerranoALMuñoz-CánovesPUnderstanding the process of fibrosis in Duchenne muscular dystrophyBiomed Res Int2014201496563124877152
- CacchiarelliDMartoneJGirardiEMicroRNAs involved in molecular circuitries relevant for the Duchenne muscular dystrophy pathogenesis are controlled by the dystrophin/nNOS pathwayCell Metab201012434135120727829
- TwayanaSLegniniICesanaMCacchiarelliDMorlandoMBozzoniIBiogenesis and function of non-coding RNAs in muscle differentiation and in Duchenne muscular dystrophyBiochem Soc Trans201341484484923863142
- Rodino-KlapacLRJanssenPMShontzKMMicro-dystrophin and follistatin co-delivery restores muscle function in aged DMD modelHum Mol Genet201322244929493723863459
- KainulainenHPapaioannouKGSilvennoinenMMyostatin/activin blocking combined with exercise reconditions skeletal muscle expression profile of mdx miceMol Cell Endocrinol201539913114225304272
- SongYHSongJLDelafontainePGodardMPThe therapeutic potential of IGF-I in skeletal muscle repairTrends Endocrinol Metab201324631031923628587
- SchertzerJDvan der PoelCShavlakadzeTGroundsMDLynchGSMuscle-specific overexpression of IGF-I improves E-C coupling in skeletal muscle fibers from dystrophic mdx miceAm J Physiol Cell Physiol20082941C161C16817989207
- SeccoMBuenoCVieiraNMSystemic delivery of human mesenchymal stromal cells combined with IGF-1 enhances muscle functional recovery in LAMA2 dy/2j dystrophic miceStem Cell Rev2013919310922664740
- PercivalJMAdamoCMBeavoJAFroehnerSCEvaluation of the therapeutic utility of phosphodiesterase 5A inhibition in the mdx mouse model of Duchenne muscular dystrophyHandb Exp Pharmacol201120432334421695647
- EnnenJPVermaMAsakuraAVascular-targeted therapies for Duchenne muscular dystrophySkelet Muscle201331923618411
- NelsonMDRaderFTangXPDE5 inhibition alleviates functional muscle ischemia in boys with Duchenne muscular dystrophyNeurology201482232085209124808022
- BertoniTJMaghASBertoniCHigh throughput screening in Duchenne muscular dystrophy: from drug discovery to functional genomicsBiology (Basel)20143475278025405319
- LongCAmoasiiLMireaultAAPostnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophyScience2016351627140040326721683
- MaggioIStefanucciLJanssenJMSelection-free gene repair after adenoviral vector transduction of designer nucleases: rescue of dystrophin synthesis in DMD muscle cell populationsNucleic Acids Res20164431449147026762977
- NelsonCEHakimCHOusteroutDGIn vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophyScience2016351627140340726721684
- OusteroutDGKabadiAMThakorePIMajorosWHReddyTEGersbachCAMultiplex CRISPR/cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophyNat Commun20156624425692716
- MendellJRRodino-KlapacLRCRISPR/Cas9 treatment for Duchenne muscular dystrophyCell Res201626551351426926391
- HeierCRDamskerJMYuQVBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effectsEMBO Mol Med20135101569158524014378
- StrehleEMStraubVRecent advances in the management of Duchenne muscular dystrophyArch Dis Child2015100121173117726153505
- VohraRSLottDMathurSMagnetic resonance assessment of hypertrophic and pseudo-hypertrophic changes in lower leg muscles of boys with Duchenne muscular dystrophy and their relationship to functional measurementsPLoS One2015106e012891526103164
- HathoutYSeolHHanMHZhangABrownKJHoffmanEPClinical utility of serum biomarkers in Duchenne muscular dystrophyClin Proteomics201613927051355
- MerliniLSabatelliPImproving clinical trial design for Duchenne muscular dystrophyBMC Neurol20151515326306629