
Abstract
Mild and moderate traumatic brain injuries (TBIs) (and concussion) occur frequently as a result of falls, automobile accidents, and sporting activities, and are a major cause of acute and chronic disability. Fatigue and excessive sleepiness are associated with increased risk of accidents, but it is unknown whether prior sleep debt also affects the pathophysiological outcome of concussive injury. Using the “dark neuron” (DN) as a marker of reversible neuronal damage, we tested the hypothesis that acute (48 hours) total sleep deprivation (TSD) and chronic sleep restriction (CSR; 10 days, 6-hour sleep/day) affect DN formation following mild TBI in the rat. TSD and CSR were administered using a walking wheel apparatus. Mild TBI was administered under anesthesia using a weight-drop impact model, and the acute neuronal response was observed without recovery. DNs were detected using standard bright-field microscopy with toluidine blue stain following appropriate tissue fixation. DN density was low under home cage and sleep deprivation control conditions (respective median DN densities, 0.14% and 0.22% of neurons), and this was unaffected by TSD alone (0.1%). Mild TBI caused significantly higher DN densities (0.76%), and this was unchanged by preexisting acute or chronic sleep debt (TSD, 0.23%; CSR, 0.7%). Thus, although sleep debt may be predicted to increase the incidence of concussive injury, the present data suggest that sleep debt does not exacerbate the resulting neuronal damage.
Introduction
Insufficient or poor-quality sleep is associated with pronounced impairments in mammalian neurobehavioral function.Citation1 In recent years, a substantial body of evidence has accumulated in support of a role for sleep in the maintenance of synaptic structure and function,Citation2 but less attention has been given to the question of whether neuronal cellular integrity is dependent on sleep and impaired by sleep deprivation.Citation3–Citation8 Two important questions arise in the latter context: i) Does sleep deprivation affect neuronal integrity or viability? ii) Does sleep deprivation influence the susceptibility of neurons to traumatic insult? Current data pertaining to both of these questions are inconclusive and inconsistent.
There is limited and contradictory evidence relating to the hypothesis that sleep deprivation can induce neuronal cell death. Markers of DNA damage and neuronal degeneration were unchanged after 5–14 days of total sleep deprivation (TSD)Citation4 or 4 days of rapid eye movement sleep deprivation (REMSD).Citation6 However, REMSD for 6 days and 10 days resulted in a temporary increase in markers of apoptosis and neuronal damage in some brainstem nuclei of rats.Citation3 Thus, REMSD may promote localized neurodegeneration but only after prolonged deprivation, and only in brainstem nuclei that are likely to be metabolically stressed by prolonged wakefulness. It is noteworthy that neurodegeneration was first observed long after the systemic physiological deficits associated with sleep loss are apparent, and it is therefore unlikely that the physiological and neurobehavioral consequences of sleep debtCitation9 are a direct result of cell death in associated neural control circuits. The rapid and apparently complete recovery of rats following prolonged TSD (even in animals exhibiting profound physiological dysfunction before the end of the procedure)Citation10 further argues against an underlying mechanism that involves permanent brain cell damage.
The above studies do not rule out the possibility that TSD may cause reversible, nonlethal changes in neuronal structure and function. The “dark neuron” (DN) is a histological marker of reversible cell damage; in the absence of ischemia or excitotoxicity, DNs are capable of recovery and begin to regain their normal staining properties in as little as 1 hour post-insult.Citation11–Citation14 There are reports in the literature that support the hypothesis that sleep deprivation can cause this kind of neuronal damage. CrileCitation15 described diffuse hyperchromatism (increased Nissl stain uptake) in cerebellar Purkinje cells following 4–5 days of TSD in rabbits and apparent reversal of this effect following recovery sleep. Eiland et alCitation5 reported argyrophilia (amino-cupric silver staining) in the supraoptic nucleus (SON) of rats after 2 days of TSD but equivocal results in other brain regions. Biswas et alCitation3 reported an increase in silver staining in some brainstem nuclei following 6 or more days of REMSD in rats.
The morphological features of DN that are visible under a light microscope include compaction (cell shrinkage), hyperbasophilia (high uptake of basic histological stains), argyrophilia (uptake of silver stains), corkscrew-like dendrites, and a triangular-shaped soma.Citation11,Citation16–Citation21 DN can form in response to a variety of physical and chemical stimuli, including (but not limited to) mechanical traumaCitation12,Citation14,Citation22,Citation23 and ischemia.Citation7,Citation24 The latter stimuli are important not only because of their relevance to pathophysiological conditions such as concussion and TBICitation25 but also because they can promote the formation of artifactual DN during tissue processing. Both mechanical trauma and global ischemia are present during autopsy and can cause DN formation in unfixed or partially fixed tissue. CammermeyerCitation17 developed a method of perfusion fixation with aldehyde fixatives that reliably produces tissue samples free of DN. This method requires that brains be perfused immediately postmortem and that autopsy be delayed for more than 24 hours. He noted that “after the lapse of 24 h the neurons are not yet quite fixed and many of them will react to trauma” (p. 249).Citation17 Formaldehyde (mainly in the form of methylene glycol) rapidly penetrates tissues and impedes autolysis, but fixation of the tissue through the formation of methylene cross-linkages is slow and may take several days at low temperature.Citation26,Citation27 Unfortunately, none of the previous studies of DN formation in sleep-deprived animalsCitation3,Citation5,Citation15 used an adequate fixation protocol, rendering their results and conclusions open to question.
There are an estimated ten million traumatic brain injuries (TBIs) per year (approximately one in every 3 seconds) worldwide.Citation28 The causes are primarily linked to falls, automobile accidents, and sporting activities, and the severity and time course of symptoms vary widely.Citation29 Causes and consequences of TBI are potentially linked to sleep in four ways: i) Sleep debt may be a risk factor for TBI. Drowsiness and excessive daytime sleepiness are risk factors for automobile and occupational accidents,Citation30,Citation31 suggesting that the growing prevalence of sleep debt might be accompanied by an increased incidence of TBI, although this has not yet been confirmed. ii) Conversely, TBI may be a risk factor for sleep dysregulation, since disruption of sleep–wake pattern is a recognized symptom of TBI and post-concussion syndrome. Sleep disturbances are reported in approximately half of young people following mild TBI and concussion.Citation32 iii) Sleep may be implicated in the recovery process following concussive or cerebrovascular brain injury,Citation33–Citation35 although remediation of sleep disturbance was not associated with improved functional outcomes.Citation36 iv) Preexisting sleep debt may influence the severity of initial damage resulting from a mild traumatic injury. The latter hypothesis has not been subjected to experimental test and is a focus of the present study.
The primary aims of the present study were to determine, with appropriate controls and tissue fixation technique, whether TSD causes DN formation in rat sensorimotor cortex, and whether sleep debt (caused by acute TSD or chronic sleep restriction [CSR]) affects the vulnerability of neurons to mild TBI.
Materials and methods
Animals and experimental protocol
All procedures conformed to guidelines of the Canadian Council on Animal Care and were approved by the animal care committee of the University of Toronto. Studies were performed using 68 male Sprague Dawley rats divided into seven groups: perfusion-fixed home cage control (HCC; n=12, body weight range 369–434 g), perfusion-fixed stimulus control (SDC; n=6, 394–435 g), immersion-fixed positive control (IMM; n=13, 351–717 g), 48 hours of TSD (n=8, 283–390 g), TBI (n=6, 339–399 g), 48 hours of TSD followed by TBI (TSD-TBI; n=13, 314–438 g), and 10 days of CSR (daily 18-hour sleep deprivation and 6-hour sleep opportunity) followed by TBI (CSR-TBI; n=10, 373–439 g).
In order to avoid inadvertent neuronal damage due to depression of the cortical surface by electrode screws, the animals were not instrumented with electroencephalographic (EEG) electrodes. Other details of the experimental protocol (laboratory setting, animal maintenance, sleep deprivation technique) prior to brain tissue autopsy and processing were as described in detail previously.Citation37 All animals were acclimatized to the laboratory setting, and a 12:12 light/dark cycle for a minimum of 1 week before the start of experiments. Animals were provided with unrestricted access to food (standard chow) and water throughout the study.
Briefly, sleep deprivation was imposed using a walking wheel revolving at 6.4 cm • s–1. The wheel was used in an intermittent rotation mode (8 seconds on and 8 seconds off cycle). In TSD, wheel rotations began at lights on (zeitgeber time, ZT0) and ended 48 hours later at the end of the dark period. In CSR, daily wheel rotations began at ZT6 (midday) and ended at the end of the dark period (ZT24). Thus, the wheel was immobile for 6 hours (ZT0–ZT6), providing a daily sleep opportunity during the early light phase. This was repeated on each of 10 consecutive days.
Mild TBI was produced using the impact injury model described and characterized by Foda and MarmarouCitation38 and Marmarou et al.Citation39 This method causes diffuse brain injury without focal lesions. Briefly, the anesthetized rat (isoflurane; 1%–2% in air/O2 mix) was placed prone with the dorsal surface of the cranium horizontal. The scalp was retracted, and an aluminum disk (10 mm diameter, 3 mm thickness) was affixed using dental acrylic cement to the surface of the cranium, centered on the sagittal suture, midway between the lambda and bregma intersections. This disk served to prevent bone fracture and subdural hematoma. A Plexiglas tube (20 mm internal diameter) was positioned vertically above the cranial disk and used as a guide for a brass projectile (impact surface 6 mm diameter). In preliminary studies, we determined that a weight of 500 g dropped from a height of 100 cm was sufficient to produce significant neuronal trauma (as indicated by DN formation) without macroscopically detectable contusive injury, and this procedure was used in all rats subjected to TBI in the present study.
Brain tissue autopsy and processing
All animals were processed in the interval ZT0–ZT6 (immediately following the end of the final sleep deprivation stimulus in TSD and CSR groups). Prior to sacrifice, animals received either an intraperitoneal dose of sodium pentobarbital (100 mg/kg) or inhalable isoflurane (1%–2% in air/O2 mix) anesthetic, followed by an intracardiac injection of 0.1 mL of heparinized (323 units/mL) saline. Since the objective of control studies was to avoid artifactual DN formation (ie, to yield brain slices with no DN), a positive control procedure was applied in every animal to confirm that histological procedures were adequate for the demonstration of DN when present. Thus, in every animal prior to sacrifice, a 0.4 mm hole was drilled in the right frontal cranium (coordinates, expressed relative to bregma, antero-posterior -2 mm, medio-lateral 2 mm) for administration of a pin prick (size 000 insect pin, diameter 0.25 mm to a depth of 4 mm) which is known to reliably cause formation of many DNs in tissue adjacent to the pin track (). Rats were then overdosed with additional anesthetic until cardiac activity ceased (analyses showed that the type of anesthetic had no statistical effect on DN counts). Brains of IMM animals were immediately carefully autopsied and submerged in a solution of 4% formaldehyde in phosphate-buffered saline (PFA; pH 7.4) at room temperature, and chilled to 4°C for at least 48 hours. All other rats were perfused transcardially with at least 100 mL of 0.9% saline, followed by 300 mL of 4% PFA for a maximum of 10 minutes. Time between cardiac arrest and start of PFA perfusion was approximately 5 minutes. Perfusates were at room temperature and were allowed to flow without restriction by gravity from 100 cm above the heart. Animals were left untouched in the supine position following PFA perfusion and maintained at 4°C for 3 days post-perfusion. Brains were then carefully autopsied and postfixed in 4% PFA at 4°C for at least an additional 24 hours.
Figure 1 ROIs used in quantitative analysis of DNs in coronal sections of the rat brain.
Abbreviations: ROI, region of interest; DN, dark neuron; Co, cortex; SON, supraoptic nucleus; Pin, cortex pin-prick perilesion.
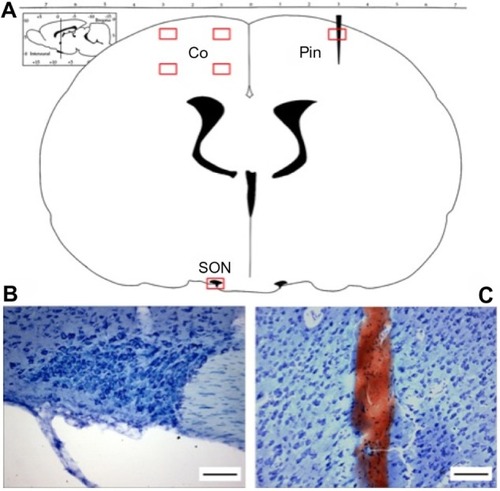
After fixation, tissues were cryoprotected with 30% sucrose in phosphate-buffered saline at 4°C for at least 48 hours, with a fresh solution change after approximately 24 hours. The brains were then flash frozen by immersion in 2-methylbutane (isopentane) kept on dry ice, and then stabilized using Tissue-Tek CRYO-O.C.T. compound (Fisher Scientific, Ottawa, Ontario, Canada). Coronal sections (bregma −0.8 mm to bregma −1.5 mm) were sliced at 40 µm (Feather blades, Fisher Scientific 12-634-1C) and mounted using 30% sucrose solution onto 3% gelatin-coated glass slides, which were allowed to dry naturally in the dark before staining. A single slice containing the SON was used for analysis of the sensorimotor cortex and SON. A separate slice containing the pin-prick track was used as a positive control for DN.
In a pilot study, three stains were evaluated: toluidine blue (TB), Nissl (cresyl violet), and silver (FD Neurosilver kit). Silver stain was found to be inconsistent; background staining was variable, and DNs were sometimes difficult to identify, even in damaged tissue (pin prick and IMM fixed). Silver staining is known to be prone to poor selectivity and reproducibility,Citation40 problems that were not alleviated by the use of a standardized kit, and silver-stained slides were therefore excluded from further analysis. Nissl and TB were found to reliably stain both normal cells and DNs. However, TB yielded lower variability of background staining and better resolution of DN, and the following analysis is therefore based only on TB-stained tissues.
A working solution of TB was prepared using 0.5 g of tolonium chloride (toluidine blue O; Sigma-Aldrich Canada Co, Oakville, ON, Canada) and 500 mL of 1% acetic acid solution. Dry slides were submerged in this 0.1% solution of TB for 40 seconds at room temperature before being well rinsed with distilled water and allowed 24 hours to dry at room temperature in the dark. Slides were then dehydrated for 2 minutes in 100% ethanol, cleared with xylene for 5 minutes, covered with Permount (Fisher Scientific Co, Ottawa, ON, Canada), and coverslipped.
Visualization of tissue preparations
Stained slides were visualized under a light microscope (BX50W1 Olympus Microscope, Carsen Group Inc., Markham, ON, Canada), and pictomicrographs were obtained using a high-resolution digital camera (3.3 RTV Micro Publisher, QIMAGING, Burnaby, BC, Canada).
Areas of the coronal slice that were selected for examination (regions of interest, ROIs) included four areas of the left sensorimotor cortex (), one ROI of the ipsilateral hypothalamus containing the SON, and one ROI of the contralateral cortex containing a pin prick as a positive control for DN. The four ROIs in the sensorimotor cortex each comprised an area of 400 µm ×600 µm, and were delineated using a standardized procedure for alignment of the field of view in every brain slice at ×40 magnification. Data from the four cortical areas were pooled for quantitative analysis. Cell counts were made at ×100 magnification ().
Figure 2 Artifactual DN formation in the cortex following immersion fixation.
Abbreviations: DN, dark neuron; HCC, perfusion-fixed home cage control; IMM, immersion-fixed positive control.
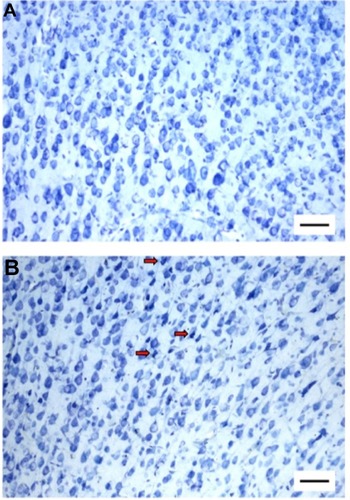
The visual identification of DN is to some extent subjective, and to minimize inconsistency, all tissue examination and cell quantification were performed by a single experimenter (AMC) using a randomized blind protocol, and using three scoring criteria: color, shape, and size. The inclusion of shape and size criteria precluded a simple automated analysis involving background subtraction and gray-scale threshold detection. Although there will undoubtedly be intra-scorer and inter-scorer variability using this approach, we assume that such variability is randomly distributed across experimental groups.
Normal neurons: appear relatively light in color with a round or teardrop shape and a size typical of the majority of neurons in the ROI.
DNs: appear dark in color with an angular shape and a compacted size in comparison to the normal neurons in the ROI.
For each ROI, the number of neurons of each type was counted using the ImageJ manual cell counter plugin (Java-based open-source software, http://rsbweb.nih.gov/ij/). Neurons touching the right edge or bottom edge of a counting grid were not included in the final cell counts, while those touching the top and left sides of the grid were included.Citation41
Particle analysis
The three criteria used in visual scoring of normal (n=723) and DN (n=126) cell types were evaluated by quantitative particle analysis (ImageJ, Java-based open-source software, http://rsbweb.nih.gov/ij/) in a sample of visually scored cells taken from positive control samples (IMM and pin-prick ROI).
Color
The color of an individual neuron was sampled from an area of the soma that did not contain the nucleus. RGB pixels were converted to brightness (Br) values using the formula Br=(R+G+B)/3, with lower values representing a darker color (value of 0 represents extreme black and 255 represents extreme white). To account for differences in staining between slides, brightness values were expressed as a percentage of normal cell brightness (averaged from a sample of 50 normal neurons on every slide).
Shape and area
Automatic edge detection was applied to 8-bit monochrome images and used to outline neurons. Cell shape (“soma form factor”, FF) was quantified as the normalized ratio of area to perimeter (FF =4πA/p2, where A is area [µm2] and p is perimeter [µm]), so that in a true circle, this ratio is one.Citation42 Values closer to zero indicate increasingly elongated shapes (eg, FF of an equilateral triangle is 0.777).
Statistical analysis
Statistical analyses were performed using Sigmastat Statistical Software (version 3.5, Systat Software Inc., Point Richmond, CA, USA) or GraphPad Prism (version 6.0f, GraphPad Software Inc., La Jolla, CA, USA). When appropriate, normally distributed homoscedastic paired data were tested using the paired t-test. In cases in which data were not normally distributed, a nonparametric Kruskal–Wallis test, with post hoc Dunn’s multiple comparisons test, was used to compare three or more groups. For two-group comparisons, the Wilcoxon signed rank test was used for matched pairs, and Mann–Whitney rank sum test was used for independent samples. Values are expressed as means ± standard error of the mean or median ± interquartile range (IQR). Effect size was quantified using the Hodges–Lehmann estimator with 95% confidence intervals (CIs). The fiducial level of statistical significance was set at P≤0.05.
Results
Quantification of morphological characteristics used as DN scoring criteria
Samples of DN from both immersion-fixed and pin-prick positive control slides were significantly less bright than normal neurons in the same slides (relative brightness 65.9%±1.27%; paired t-test, P<0.001). DNs were significantly less circular (more elongated) than normal neurons (FF, median [IQR], 0.58 [0.55–0.61] and 0.68 [0.63–0.73], in DNs and normal neurons, respectively; Wilcoxon signed rank test, P<0.001). The area of DN was significantly smaller than normal neurons (98.0 [92.4–106.5] and 159.5 [146.1–184.1] µm2, respectively; Wilcoxon signed rank test, P=0.001).
Positive and negative controls
The cortex of perfusion-fixed brains autopsied from HCC animals had very few DNs (and usually none; median [IQR], 0.14 [0.07–0.27]% of neurons in ROI). Likewise, SDC rats exhibited very low DN counts (0.22 [0.10–0.40]%). These controls were not statistically significantly different (Mann–Whitney test, P=0.45). DNs were observed surrounding the cortical pin-prick lesion in every case (), thereby validating the tissue preparation and histological techniques and confirming that DN-free samples were legitimate. In contrast, despite taking great care during immersion fixation procedures, the brains of the IMM group contained appreciable numbers of DNs (1.41 [0.78–2.21]%) that were randomly dispersed among normal neurons (), confirming that incompletely fixed tissue is susceptible to artifactual DN formation during autopsy.Citation17,Citation20
There was no difference in the number of DNs in HCC animals anesthetized with pentobarbital or isoflurane (unpaired t-test, P=0.283).
Supraoptic nucleus
Compacted DN could not be identified in the SON of any animal, including animals whose brains were immersion-fixed (in which DNs were readily visible in the cortex). To increase the probability of forming DNs in the SON, six of the IMM animals were subjected to intentional trauma in the approximate region of the SON, while the brain was intact and unfixed. A brass weight of either 1 g or 50 g was placed on the ventral surface of the brain for 2 minutes prior to immersion fixation in 4% PFA. Despite this mechanical insult, DNs could not be distinguished from normal neurons in the SON, even when DNs were conspicuous in hypothalamic areas adjacent to the SON.
Neurons of the SON stained more darkly (Br=84.3±5.1) than normal cortical neurons (Br=111.0±7.7) (paired t-test, P=0.007). Cell area and FF of SON neurons were equivalent in HCC and IMM (P=0.11 and 0.89 for area and FF, respectively). Thus, all three of the criteria used in this study failed to discriminate between normal cells and DNs in the SON. SON data were therefore excluded from further analysis.
Cortex: effect of TSD
Forty-eight hours of TSD in the walking wheel apparatus produced no significant difference in cortex DN density (0.10 [0.08–0.32]%) compared to either HCC or SDC animals (Kruskal–Wallis test, P=0.623; ). Hodges–Lehmann estimator was 0 [95% CI, −0.1566 to 0.07828] for HCC vs TSD and -0.078 [−0.3131 to 0.2740] for SDC vs TSD.
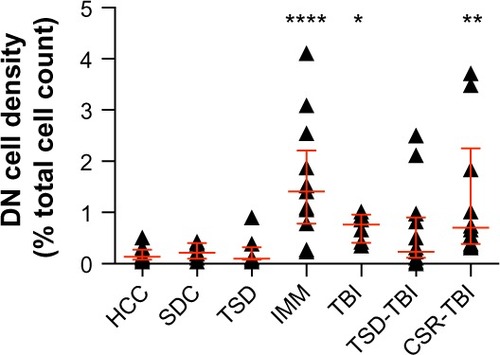
Figure 3 DN counts in rat sensorimotor cortex.
Abbreviations: DN, dark neuron; HCC, perfusion-fixed home cage control; SDC, perfusion-fixed stimulus control; TSD, total sleep deprivation; IMM, immersion-fixed positive control; TBI, traumatic brain injury; TSD-TBI, TSD followed by TBI; CSR-TBI, chronic sleep restriction followed by TBI.
Cortex: effect of TBI, with and without prior sleep debt
Induction of mild TBI by the weight-drop protocol successfully caused increased formation of DN in the cortex; DN density was significantly greater in the TBI group (0.76 [0.41–0.96]%) than in HCC and SDC groups (Kruskal–Wallis test, P<0.0001), confirmed by Hodges–Lehmann estimators (0.587 [0.274–0.822] and 0.489 [0.117–0.822] for TBI vs HCC and TBI vs SDC, respectively). Prior sleep debt induced by 48 hours of TSD or 10 days of CSR had no statistically significant effect on DN density vs TBI alone (TSD-TBI, 0.23 [0.12–0.90]%; CSR-TBI, 0.70 [0.38–2.25]%; Kruskal–Wallis test, P=0.083; ). The absence of effect was supported by Hodges–Lehmann estimators, which overlapped zero in both TSD-TBI vs TBI (−0.313 [−0.44 to 0.391]) and CSR-TBI vs TBI (0.059 [−0.391 to 2.544]). However, a prominent feature of the responses to TBI was a higher inter-animal variability in DN density compared with controls (TBI vs HCC groups in ). Prior sleep debt appeared to increase this variability (TBI vs TSD-TBI and CSR-TBI groups in ), but this effect could not be evaluated statistically.
Discussion
The present study shows for the first time that preexisting sleep debt does not affect the vulnerability of cortical sensorimotor neurons to the immediate effects of mild TBI. We also refute the hypothesis that acute (48 hours) TSD causes DN formation in the sensorimotor cortex of the rat, corroborating conclusions reached in previous research.Citation3,Citation5
In a previously published study on the effect of TSD on DN formation,Citation5 rat brains were processed or manipulated in less than 24 hours following perfusion of formaldehyde fixative, which is known to cause artifactual formation of DN.Citation16–Citation18 Some of the animals in that studyCitation5 were described as exhibiting “a diffuse, dust-like reactive staining … in a wide area of dorsal cortex”, but a statistically significant effect of TSD could not be demonstrated.Citation5 The results of the present study are consistent with this negative statistical conclusion and reinforce the suggestion that the above data may have been compromised by inappropriate tissue-handling technique. Samples taken from the IMM group in this study show that this issue is not trivial, since DN counts due to careful autopsy of unfixed brain were at least as great as those caused by impact trauma in the TBI group (). In contrast, by delaying autopsy for 3 days after perfusion fixation, control group brains (HCC and SDC) were shown to contain very few or no DN ( and ). The positive controls (pin prick) confirmed this to be a genuine observation of absence, and not faulty technique.
Biswas et alCitation3 observed argyrophilia in brainstem nuclei under all conditions, including control, but nevertheless reported significantly greater DN in rats subjected to prolonged REMSD (≥6 days, by the flowerpot technique) than in control animals. Notably, the enhancement of argyrophilia was absent following 3 days of subsequent sleep recovery. However, the inadequate fixation technique used in that study admits two possible conclusions: that REMSD was the direct cause of neuronal damage and that prolonged REMSD enhanced the susceptibility of brain to artifactual damage. The present study does not support the latter interpretation, since TBI did not cause higher DN counts in animals with sleep debt (compared with those subjected to TBI without prior TSD or CSR).
Eiland et alCitation5 examined whole-brain argyrophilia following prolonged TSD (8–10 days using the disk-over-water technique). Argyrophilia was observed in all animals, but a statistically significant difference between TSD and control was found only in the SON. Unfortunately, our attempts to verify this result were unsuccessful because we found that DN could not be reliably identified in SON using the TB and Nissl stains. The magnocellular neurosecretory cells of the SON contain large quantities of rough endoplasmic reticulum, which is basophilic, and therefore, the SON neurons stain darkly even under control conditions (). Eiland et alCitation5 reported the same result using Nissl stain. We found no evidence of cell compaction (reduced size and circularity) in SON neurons, even in IMM samples or following application of pressure to the ventral surface of the unfixed brain. This failure of the positive control precludes a conclusion, and further research will be required to confirm or refute the report by Eiland et al.Citation5
In a previous study,Citation37 we found that EEG- and electromyogram (EMG)-instrumented rats subjected to 48 hours of TSD and 10 days of CSR accumulated sleep deficits of 21 hours and 55 hours, respectively, and we assume similar sleep deficits under identical conditions in the TSD and CSR groups of uninstrumented rats in the present study. This level of sleep debt, while not severe, is nevertheless sufficient to elicit appreciable neurobehavioral and physiological effects. In rats, TSD of 1–4 days is associated with a progressive suppression of behavioral and EEG sleep–wake homeostatic responsesCitation43 and a rise in body temperature and metabolic rate.Citation9 Acute TSD also affects neurobehavioral performance, learning and memory, and changes in synaptic plasticity and neuronal proliferation.Citation1 Taken together, these data suggest that 48-hour TSD is a significant stressor on central nervous system and systemic function and is consistent with a hypothesis that TSD can compromise neuronal function. Two recent studies of young athletes are of particular relevance in this context because they show that the scores obtained in standard tests used in the evaluation of neurocognitive symptoms of concussion are sensitive to prior sleep history in baseline (non-concussed) subjects.Citation44,Citation45 The CSR protocol has also been found to induce long-term alterations in sleep homeostasis in rats,Citation46,Citation47 as well as memory and neurobehavioral impairment and effects on immune and peripheral tissue function.Citation48–Citation52 Many of the above-mentioned consequences of sleep deprivation are rapidly reversible upon resumption of unrestricted sleep, suggesting that the mechanisms involved in any underlying neuronal dysfunction are reversible. Studies have shown that widespread neuronal degeneration through necrotic and apoptotic mechanisms is not apparent during even prolonged TSD,Citation4,Citation6 prompting us to search for signs of an alternative form of neuronal structural impairment as represented by the DN. The data did not support this hypothesis, and we therefore conclude that the mechanisms leading to DN compaction and hyperbasophilia are unaffected by moderate sleep debt in rat cortex.
Furthermore, we show for the first time that the susceptibility of the mechanism leading to DN in response to mild TBI is not strongly affected by acute and chronic sleep debt. We confirmed that the impact model was effective in causing neuronal damage (as reflected by DN density) in the cortex immediately below the point of impact, and we focused our attention on this brain region. A more comprehensive survey of other brain regions may reveal differential effects, but this was not undertaken in the present study. demonstrates that acute and chronic sleep debt did not increase the median DN density following TBI. This was confirmed by the 95% CIs of the Hodges–Lehmann estimator, which shows clearly that the small effect size of TSD-TBI and CSR-TBI, relative to TBI alone, cannot be statistically distinguished from zero. We calculate (Statistical Solutions, LLC, Cottage Grove, WI, USA; http://www.statisticalsolutions.net/pss_calc.php) using the HCC data (DN density mean ± standard deviation, 0.1761±0.1411, n=12), that the sample size needed to detect a doubling of DN density at statistical power 0.8 is n=6. We sampled n=8, 6, 13, and 10 for TSD, TBI, TSD-TBI and CSR-TBI groups, respectively. For these reasons, we conclude that the negative result is genuine and is unlikely to reflect a type II statistical error. In this context, it is important to note that prior sleep debt (both TSD and CSR) increased the variance of the response to TBI. This implies that prior sleep debt had a greater effect on some animals than others, the reasons for which are unclear but might relate to different levels of sleep debt achieved. Unfortunately, we did not record sleep in this study (for reasons given earlier), and are unable to evaluate this possibility.
This negative result is consistent with a recent finding that baseline (pre-TBI) sleep is a poor predictor of post- concussion symptoms in collegiate athletes.Citation53 Three studiesCitation7,Citation35,Citation54 in rodents have tested the hypothesis that prior sleep debt influences the severity of neuronal damage resulting from ischemia (stroke), and a fourth study examined the effect of sleep debt on excitotoxicity.Citation55 All four studies hypothesized that loss of sleep would worsen the impact of the insult but instead found that sleep debt appeared to protect the tissues through an unknown “preconditioning” mechanism. There were considerable differences between studies in the magnitude of pre-insult sleep debt and the methods used to achieve it, ranging from a brief 6-hour TSD by gentle handling in ratsCitation54 to 2 days of mainly REMSD using the platform technique in mice,Citation35 to 5 days of TSD by the disk-over-water technique in rats,Citation7 and 30 days of CSR (4-hour sleep opportunity per day) using a rotating drum.Citation55 The consistency of the result is compelling in view of this variation in stimulus. Our data, which examined the effects of non-contusive impact injury, further support the conclusion that preexisting sleep debt does not worsen the immediate consequences of brain injury. However, we did not find evidence that sleep debt suppressed the TBI-mediated formation of DN. An important difference between the present study and those prior studies concerns the experimental timeline. Specifically, we examined the acute effect of TBI by processing the animals immediately after application of the impact, whereas all four of the prior studies allowed 7–8 days of recovery before the animals were killed and brains processed. Those studies therefore examined the combined effects of sleep debt on neuronal susceptibility to insult and potential delayed damage, plus the capacity to recover over the 1st week. It is possible that postimpact neuronal damage may develop over time in our model of mild TBI, especially if the trauma is associated with undetected cerebrovascular disturbance or cellular energy crisis.Citation29 However, the present study was concerned only with the immediate susceptibility of the neurons to impact, which was the principal reason for our choice of the DN as a marker of cellular damage. It is well known that DN formation is very rapid, possibly involving a biophysical mechanism of gel–gel phase transition.Citation11,Citation56 Indeed, we placed much emphasis on appropriate fixation techniques precisely because the DN formation process is rapid enough to occur within the time needed for complete fixation of brain tissue using PFA fixatives.Citation16–Citation18 The positive controls (pin-prick trauma) demonstrated in each case that DN formation was not time limited in our protocol. Thus, we have shown that the immediate neuronal effects of impact are minimally affected by sleep debt, but we cannot exclude the possibility that sleep debt might influence delayed effects. For example, it is possible that sleep debt might confer a protective function in mild TBI by increasing the proportion of cells that recover, a hypothesis that deserves further study.
Conclusion
Excess sleepiness and sleep deprivation are associated with increased risk of accident,Citation30,Citation31 but the present study has shown that sleep deprivation does not worsen the attendant neuronal damage immediately following concussive injury. Furthermore, by taking care to avoid the artifactual formation of DNs, we have confirmed that acute sleep deprivation does not induce neuronal degeneration in otherwise healthy rat cortex. Thus, sleep debt neither causes neuronal cell damage nor exacerbates damage arising from mild traumatic injury.
Acknowledgments
This study was funded by the Natural Sciences and Research Council of Canada (NSERC) through a Discovery Grant to RS and Postgraduate Scholarship to AMC. NSERC had no involvement in the design, conduct, or interpretation of this study.
We are grateful to Henry Hong for technical assistance and advice, and to Dr John Peever for the use of his microscope and associated equipment.
Disclosure
Both authors declare no conflict of interest.
References
- McCoyJGStreckerREThe cognitive cost of sleep lostNeurobiol Learn Mem201196456458221875679
- RibeiroSSleep and plasticityPflugers Arch2012463111112021947578
- BiswasSMishraPMallickBNIncreased apoptosis in rat brain after rapid eye movement sleep lossNeuroscience2006142231533116887278
- CirelliCShawPJRechtschaffenATononiGNo evidence of brain cell degeneration after long-term sleep deprivation in ratsBrain Res19998401–218419310517970
- EilandMMRamanathanLGulyaniSIncreases in amino-cupric-silver staining of the supraoptic nucleus after sleep deprivationBrain Res200294511812113945
- HipolideDCD’AlmeidaVRaymondRTufikSNobregaJNSleep deprivation does not affect indices of necrosis or apoptosis in rat brainInt J Neurosci2002112215516612325404
- HsuJCLeeYSChangCNLingEALanCTSleep deprivation prior to transient global cerebral ischemia attenuates glial reaction in the rat hippocampal formationBrain Res20039841–217018112932851
- MorrisseyMJDuntleySPAnchAMNonnemanRActive sleep and its role in the prevention of apoptosis in the developing brainMed Hypotheses200462687687915142640
- RechtschaffenABergmannBMSleep deprivation in the rat: an update of the 1989 paperSleep2002251182411833856
- EversonCAGillilandMAKushidaCASleep deprivation in the rat: IX. RecoverySleep198912160672538911
- CsordasAMazloMGallyasFRecovery versus death of “dark” (compacted) neurons in non-impaired parenchymal environment: light and electron microscopic observationsActa Neuropathol20031061374912665989
- KheraniZSAuerRNPharmacologic analysis of the mechanism of dark neuron production in cerebral cortexActa Neuropathol2008116444745218521615
- KovesdiEPalJGallyasFThe fate of “dark” neurons produced by transient focal cerebral ischemia in a non-necrotic and non-excitotoxic environment: neurobiological aspectsBrain Res2007114727228317349980
- OoigawaHNawashiroHFukuiSThe fate of Nissl-stained dark neurons following traumatic brain injury in rats: difference between neocortex and hippocampus regarding survival rateActa Neuropathol2006112447148116858608
- CrileGWStudies in exhaustion: an experimental researchArch Surg192122196220
- CammermeyerJThe post-mortem origin and mechanism of neuronal hyperchromatosis and nuclear pyknosisExp Neurol1960237940513807188
- CammermeyerJThe importance of avoiding “dark” neurons in experimental neuropathologyActa Neuropathol19611245270
- CammermeyerJIs the solitary dark neuron a manifestation of postmortem trauma to the brain inadequately fixed by perfusion?Histochemistry19785629711597249
- GallyasFGuldnerFHZoltayGWolffJRGolgi-like demonstration of “dark” neurons with an argyrophil III method for experimental neuropathologyActa Neuropathol19907966206281694382
- JortnerBSThe return of the dark neuron. A histological artifact complicating contemporary neurotoxicologic evaluationNeurotoxicology200627462863416650476
- ZsombokATothZGallyasFBasophilia, acidophilia and argyrophilia of “dark” (compacted) neurons during their formation, recovery or death in an otherwise undamaged environmentJ Neurosci Methods2005142114515215652628
- GallyasFZoltayGAn immediate light microscopic response of neuronal somata, dendrites and axons to non-contusing concussive head injury in the ratActa Neuropathol19928343863931374204
- GallyasFZoltayGBalasIAn immediate light microscopic response of neuronal somata, dendrites and axons to contusing concussive head injury in the ratActa Neuropathol19928343944011374205
- CzurkoANishinoH‘Collapsed’ (argyrophilic, dark) neurons in rat model of transient focal cerebral ischemiaNeurosci Lett19931621–271748121640
- GoldingEMRobertsonCSBryanRMJrThe consequences of traumatic brain injury on cerebral blood flow and autoregulation: a reviewClin Exp Hypertens199921429933210369378
- FoxCHJohnsonFBWhitingJRollerPPFormaldehyde fixationBiotech Histochem1985338845853
- KiernanJAFixation of mast cellsBiotech Histochem2005801444516041888
- HyderAAWunderlichCAPuvanachandraPGururajGKobusingyeOCThe impact of traumatic brain injuries: a global perspectiveNeuroRehabilitation200722534135318162698
- PrinsMGrecoTAlexanderDGizaCCThe pathophysiology of traumatic brain injury at a glanceDis Model Mech2013661307131524046353
- StrohlKPBrownDBCollopNATS Ad Hoc Committee on Sleep Apnea, Sleepiness, and Driving Risk in Noncommercial Drivers. An official American Thoracic Society Clinical Practice Guideline: sleep apnea, sleepiness, and driving risk in noncommercial drivers. An update of a 1994 StatementAm J Respir Crit Care Med2013187111259126623725615
- UehliKMehtaAJMiedingerDSleep problems and work injuries: a systematic review and meta-analysisSleep Med Rev2014181617323702220
- BarlowKMPostconcussion syndrome: a reviewJ Child Neurol2014291111
- GaoBCamEJaegerHZunzuneguiCSarntheinJBassettiCLSleep disruption aggravates focal cerebral ischemia in the ratSleep201033787988720614848
- Martinez-VargasMEstrada RojoFTabla-RamonESleep deprivation has a neuroprotective role in a traumatic brain injury of the ratNeurosci Lett2012529211812223022503
- WeilZMNormanGJKarelinaKSleep deprivation attenuates inflammatory responses and ischemic cell deathExp Neurol2009218112913619409382
- CastriottaRJAtanasovSWildeMCMaselBELaiJMKunaSTTreatment of sleep disorders after traumatic brain injuryJ Clin Sleep Med20095213714419968047
- CaronAMStephensonREnergy expenditure is affected by rate of accumulation of sleep deficit in ratsSleep20103391226123520857870
- FodaMAMarmarouAA new model of diffuse brain injury in rats. Part II: morphological characterizationJ Neurosurg19948023013138283270
- MarmarouAFodaMAvan den BrinkWCampbellJKitaHDemetriadouKA new model of diffuse brain injury in rats. Part I: pathophysiology and biomechanicsJ Neurosurg19948022913008283269
- GallyasFPhysicochemical mechanisms of histological silver staining and their utilization for rendering individual silver methods selective and reliableBiotech Histochem200883522123819016367
- GundersenHJBaggerPBendtsenTFThe new stereological tools: disector, fractionator, nucleator and point sampled intercepts and their use in pathological research and diagnosisAPMIS198896108578813056461
- JinnoSKinukawaNKosakaTMorphometric multivariate analysis of GABAergic neurons containing calretinin and neuronal nitric oxide synthase in the mouse hippocampusBrain Res2001900219520411334798
- RechtschaffenABergmannBMGillilandMABauerKEffects of method, duration, and sleep stage on rebounds from sleep deprivation in the ratSleep199922111319989363
- McClureDJZuckermanSLKutscherSJGregoryAJSolomonGSBaseline neurocognitive testing in sports-related concussions: the importance of a prior night’s sleepAm J Sports Med201442247247824256713
- MihalikJPLengasERegister-MihalikJKOyamaSBegalleRLGuskiewiczKMThe effects of sleep quality and sleep quantity on concussion baseline assessmentClin J Sport Med201323534334823917732
- KimYLaposkyADBergmannBMTurekFWRepeated sleep restriction in rats leads to homeostatic and allostatic responses during recovery sleepProc Natl Acad Sci U S A200710425106971070217548824
- StephensonRCaronAMFaminaSBehavioral sleep-wake homeostasis and EEG delta power are decoupled by chronic sleep restriction in the ratSleep201538568569725669184
- DeurveilherSBushJERusakBEskesGASembaKPsychomotor vigilance task performance during and following chronic sleep restriction in ratsSleep201538451552825515100
- EversonCAHenchenCJSzaboAHoggNCell injury and repair resulting from sleep loss and sleep recovery in laboratory ratsSleep201437121929194025325492
- McCoyJGChristieMAKimYChronic sleep restriction impairs spatial memory in ratsNeuroreport2013242919523238166
- Van DongenHPMaislinGMullingtonJMDingesDFThe cumulative cost of additional wakefulness: dose-response effects on neurobehavioral functions and sleep physiology from chronic sleep restriction and total sleep deprivationSleep200326211712612683469
- ZagerAAndersenMLRuizFSAntunesIBTufikSEffects of acute and chronic sleep loss on immune modulation of ratsAm J Physiol Regul Integr Comp Physiol20072931R504R50917409265
- MerrittVCArnettPAPremorbid predictors of postconcussion symptoms in collegiate athletesJ Clin Exp Neuropsychol201436101098111125493542
- MoldovanMConstantinescuAOBalseanuAOprescuNZagreanLPopa-WagnerASleep deprivation attenuates experimental stroke severity in ratsExp Neurol2010222113514320045410
- NovatiAHulshofHJGranicIMeerloPChronic partial sleep deprivation reduces brain sensitivity to glutamate N-methyl-D-aspartate receptor-mediated neurotoxicityJ Sleep Res20122113921672070
- GallyasFPalJBukovicsPSupravital microwave experiments support that the formation of “dark” neurons is propelled by phase transition in an intracellular gel systemBrain Res2009127015215619324025
- PaxinosGWatsonCThe Rat Brain in Stereotaxic Coordinates4th edSan Diego, CAAcademic Press1998