Abstract
The study of complex genetics in autoimmune diseases has progressed at a tremendous pace over the last 4 years, as a direct result of the enormous gains made by genome wide association studies (GWAS). Novel genetic findings are continuously being reported alongside the rapid development of genetic technologies, sophisticated statistical analysis, and larger sample collections. It is now becoming clear that multiple genes contribute to disease risk in many complex genetic disorders including rheumatoid arthritis (RA) and that there are common genetic risk factors that underlie a spectrum of autoimmune diseases. This review details the current genetic landscape of RA, and describes what GWAS has taught us in terms of missing heritability, subsets of disease, existence of genetic heterogeneity, and shared autoimmune risk loci. Finally, this review addresses the initial challenges faced in translating the wealth of genetic findings into determining the biological mechanisms that contribute to the relationship between genotype and phenotype. Unraveling the mechanism of how genes directly influence the cause of RA will lead to a better understanding of the disease and will ultimately have a direct clinical impact, informing the development of new therapies that can be utilized in the treatment of RA.
Introduction
Rheumatoid arthritis (RA) is a complex, chronic autoimmune disease (AID) that affects approximately 0.5%–1% of the population worldwide. It is characterized by inflammation and progressive destruction of the synovial joints leading to pain, long-term disability, and reduced quality of life in many patients.Citation1 Although RA primarily affects joints it can also result in extra-articular disease manifestations, which tend to occur in patients with more severe disease.Citation2 According to most epidemiological studies, RA is more prevalent in women and the peak age of onset is typically around the fifth decade, although there is evidence to suggest this is rising.Citation3 There are significant variations of disease incidence and prevalence amongst different populations, suggesting an association of RA with ethnicity.Citation4 RA continues to impose a substantial economic burden on society resulting from the significant morbidity and premature mortality associated with the disease, with the life expectancy of RA patients shortened by 3–10 years.Citation5
The etiology of RA remains largely unknown, although it is established as a multifactorial disease resulting from a complex interplay between genetic and environmental factors. A genetic link to RA was first established through the observation of familial clustering in cases,Citation6 siblings of affected patients having an increased relative risk of RA compared to the general population (λs), ranging between 5 and 10.Citation7 Twin studies have also provided compelling evidence to support this genetic component, with disease concordance between monozygotic twins (15%) being considerably higher compared to dizygotic twins (3.6%).Citation8,Citation9 From such studies, the overall heritability of RA has been estimated to be between 50% and 60%.Citation10 It is documented that genetic risk factors contribute to both occurrence and severity of disease.Citation11 Low concordance rates between monozygotic twins emphasize the importance of environmental factors in RA susceptibility. Long-term smoking remains the only validated environmental factor that contributes to an increased risk of developing seropositive RA,Citation12 although studies have implicated other potential environmental risk factors including hormones, pollution, diet, and infectious agents.Citation13–Citation15 Ongoing work will continue to evaluate the role of environmental factors and place importance on dissecting gene–environment (G–E) interactions.
Genetics of RA
The need to uncover the full genetic risk component of RA is imperative for improving understanding of the disease and may inform the development of new therapies, improved diagnosis, prevention, and potentially prediction of disease risk and severity.
The last 60 years have seen mounting evidence to support the genetic basis of RA through the identification of genetic susceptibility variants. Three main approaches have been used to identify these susceptibility loci: candidate gene, linkage, and genome wide association studies (GWAS), the third being the most successful. The latest GWAS meta-analysis association study has brought the total number of confirmed RA risk loci to 34.Citation16 A comprehensive discussion of all the identified genetic loci is beyond the scope of this review; however the major landmarks in the search of susceptibility loci will be covered. illustrates all confirmed RA loci to date in approximate order of discovery complete with heritability estimates.
Figure 1 Current rheumatoid arthritis (RA) genetic risk loci. Each confirmed RA risk locus has been plotted in order of approximate discovery from left to right on the bottom axis. For each locus the odds ratio and confidence interval of the most significant allele has been plotted against the y-axis (left). On the y-axis (right) the increase in heritability explained by confirmed loci has been plotted (λs = 5). * validated in east-Asian populations only.
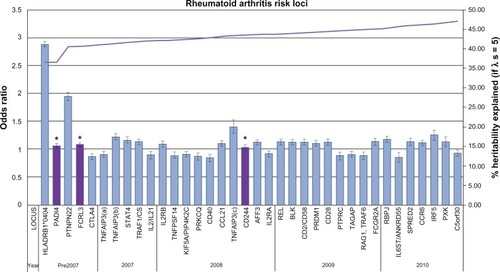
Prior to the development of GWAS, RA susceptibility loci were discovered through candidate gene and linkage studies. Although these approaches led to the discovery of only a small number of loci, they identified the two most significant RA risk loci that remain to date: human leukocyte antigen (HLA-DRB1) and protein tyrosine phosphatase, nonreceptor type 22 (PTPN22). Together HLA-DRB1 and PTPN22 are estimated to account for approximately 40% of the total genetic risk for RA.Citation17 More recently these approaches have also led to the identification of the signal transducer and activator of transcription 4 (STAT4) locus.
HLA-DRB1
The HLA region remains the most statistically significant and important genetic association for RA. The knowledge of a marked involvement of HLA-DRB1 with the immune response identified it as a good candidate gene for RA. Stastny first demonstrated an association of HLA-DRw4 with RA in the 1970s using serological techniques in population studies.Citation18 This initial finding evolved over time to include association of several distinct alleles of the class II gene, HLA-DRB1. Molecular typing techniques were developed to investigate the complexity of the locus and work by Gregersen et al led to the observation that the associated alleles encode for a conserved amino acid sequence in the third hypervariable region of the DRB1 chain.Citation19 Subsequently this region was termed the shared epitope (SE). The SE hypothesis is based on the theory that these class II alleles are directly involved in the pathogenesis of RA. The molecular basis of this association remains elusive but present models suggest that structural differences between alleles could be involved at the level of antigen presentation or in the alteration of the T cell repertoire.Citation20
The alleles in the HLA-DRB1 locus have been consistently associated with RA in many populations through linkage and association studies despite the fact that SE allele frequencies differ significantly between ethnic groups. More recently, studies have shown evidence of association for additional RA risk alleles within the major histocompatibility complex (MHC) region, adding further complexity,Citation21 although the extended linkage disequilibrium (LD) across this region makes interpretation of these associations very difficult.Citation22 Ongoing work has led to suggestions that MHC associations with RA cannot wholly be accounted for by the SE hypothesis and there have been several attempts to modify and redefine this concept.Citation23,Citation24 Although the MHC locus confers the largest risk for RA susceptibility, it is neither necessary nor sufficient to cause disease.
PTPN22
PTPN22 was the first locus outside the MHC to demonstrate consistently validated association with RA. PTPN22 is recognized as the second most important risk loci to the MHC in populations of European descent. The minor allele of a nonsynonymous single nucleotide polymorphism (SNP), (rs2476601, 1858 C > T, R620W) in PTPN22 was first associated with type 1 diabetes (T1D) in a candidate gene study.Citation25 This variant was subsequently confirmed at a similar time in RA through assaying 87 putative functional SNPs in RA candidate genes and/or linkage regions.Citation26 Since then, the association with PTPN22 has been replicated in several other populations,Citation27–Citation34 and the same variant is also associated with a variety of other autoimmune and inflammatory diseases.Citation35–Citation39 Interestingly no variants within this gene have been confirmed to be associated with RA in Asian populations where the second largest RA genetic risk factor is recognized as PADI4.Citation40 The PTPN22 gene is a compelling biological candidate being a key component in the regulation of T cell receptor signaling.Citation25 The associated risk allele encodes an arginine to tryptophan substitution at residue 620 in the polypeptide chain disrupting the binding to Csk. There is evidence that this change confers increased function to the PTPN22 protein, with the 620W variant enhancing the inhibitory effect on T cell receptor signaling.Citation41
STAT4
The association of STAT4 with RA was discovered through a combination of linkage and candidate gene studies. In contrast with HLA-DRB1 and PTPN22, the association of STAT4 with RA is more modest. This locus was identified using a fine mapping approach to examine 13 candidate genes previously found under a peak of linkage identified on chromosome 2q.Citation42 STAT4 is an example of a loci that confers susceptibility to RA across all populations tested to date as well as being associated with a variety of other AIDs.Citation43–Citation46 STAT4 itself is part of a family of seven transcription factors involved in cytokine receptor signaling, initially activated by IL-12 signaling in T cells, and then phosphorylated by janus kinases. Subsequently, STAT4 is translocated to the cell nucleus where it initiates transcription of its target genes and results in the production of interferon-γ and Th1 responses. Transgenic knockout mice for STAT4 have proved that it is critical to the development and function of T helper cells. Defective mice are resistant to proteoglycan-induced arthritis, do not respond to IL-12, and lack Th1 responses.Citation47 Although rodent models of arthritis do not perfectly duplicate the clinical pathology of RA they have demonstrated good predictability and efficacy exemplified by the successful development of therapeutics for clinical use.Citation48
The use of traditional linkage and candidate gene studies to uncover genetic susceptibility has not led to definitive answers regarding the etiology of RA. The limited success in the application of linkage analysis to complex diseases is predominately due to the inadequate power and resolution to detect variants of modest effect.Citation49 Candidate gene studies have in the past resulted in numerous associations that are difficult to replicate, and in some cases, to overestimated genetic effects.Citation50 This approach is also limited by its reliance on prior knowledge of the biology of the disease and by the highly subjective selection of potential genes. The alternative approach to these methods are GWAS which entail a systematic search of the entire genome for susceptibility variants. Over the past 4 years, the implementation of GWAS has brought tremendous gains to knowledge in many complex diseases.
GWAS
GWAS have become the most powerful and extensively employed approach in discovering susceptibility variants for complex disease traits. Advances in technology, reduced costs, and the ascertainment of large well-characterized case cohorts and panels of controls have made GWA scans widely available. GWAS have also been made possible through several other major advances in the last decade. The completion of the Human Genome Project in 2001Citation51 and the initial release of the International Hap Map project in 2003Citation52 paved the way forward for this approach, allowing the design of genotyping arrays to capture variation across the entire genome. These studies are based on genetic association, determined if genetic variants are seen more or less frequently in disease affected cohorts compared to non affected cohorts in the same population. The genetic markers typically used in these studies are SNPs, as they are abundant and easy to genotype. The SNP genotyping arrays, however, have very limited coverage and power to detect rare variation associated to disease, so the underlying rationale for GWAS is the common disease common variant hypothesis (CDCV).Citation53 This proposes that common disease alleles (SNPs) with a minor allele frequency (MAF) >1% underlie most common diseases.Citation54
The success and popularity of this strategy has been marked, evidenced by an online catalog which contained 812 published GWAS for 476 different complex traits as of March 2011, and is still growing.Citation55 As with other complex diseases, GWAS has expanded the number of confirmed RA susceptibility loci dramatically with 34 genetic loci now confidently associated (). A summary of these findings gives an insight to the current genetic landscape of RA.
In the same year that STAT4 was identified as a risk locus for RA the first major GWAS was conducted by the Wellcome Trust Case-Control Consortium (WTCCC). The WTCCC is an assembly of 50 research groups across the UK established in 2005. Its primary purpose is to accelerate efforts in deciphering the human genome sequence variation and to identify variants responsible for the major causes of morbidity and mortality.
WTCCC – GWAS
In 2007, a revolutionary GWAS was undertaken by the WTCCC in 14,000 cases of seven common diseases, including 2000 United Kingdom RA cases collected by the Arthritis Research United Kingdom Epidemiology Unit and ∼3000 shared controls. This was the largest single GWAS of its time. Cases and controls were genotyped using the Affymetrix Gene Chip (Santa Clara, CA) 500 k array.Citation56 The success of this study is demonstrated by the identification of 24 independently associated regions across all seven diseases. Association with RA at genome-wide significance level (P < 5 × 10−8) was validated at the robustly associated susceptibility loci: HLA-DRB1 and PTPN22. Nine other loci were associated in the next tier of significance (Tier 2, P = 1 × 10−5 − 5 × 10−7) none of which had been previously associated with RA. In Tier 3, 49 SNPs were identified with modest association with RA (P = 1 × 10−4 − 1 × 10−5). Many of these suggestive associations have subsequently been validated as RA susceptibility loci in replication studies.
Since the first WTCCC GWAS, many different strategies have been used in attempting to identify additional RA risk loci. These include validation studies, further GWAS, meta-analysis studies, candidate gene studies, and bioinformatics approaches.
GWAS validation studies
With the extensive number of significant associations being reported through GWAS, it is imperative that the findings are validated through replication in large independent cohorts and potentially confirmed in other populations to ensure that they are real findings. GWAS validation studies have been employed extensively since the first WTCCC study. Many follow-up studies were initiated for the Tier 2 and Tier 3 WTCCC SNPs, and as a result, confirmed numerous novel associations. The 6q23 locus provides a good example of the use of this strategy. Following the WTCCC GWAS, Thomson et al followed up nine RA Tier 2 SNPs in a large independent UK cohort of 5063 cases and 3849 controls.Citation57 One of these SNPs, rs6920220, was replicated reaching a level of genome wide significance (P = 1.1 × 10−8). Simultaneously, this SNP and a second independent polymorphism was detected in the 6q23 region in a GWAS of a US population which confirmed its importance as a risk loci for RA.Citation58 Fine mapping of this region has revealed the complexity of this locus which has at least three independent effects. Two of these alleles, rs6920220 and rs5029937, confer risk and one, rs13207033, confers protection to RA. Also, two of the variants map to an intergenic region between OLIG3 and TNFAIP3 and one (rs5029937) maps to the second intron of TNFAIP3i.Citation59 Since OLIG3 is involved in the development and differentiation of neuronal cells, TNFAIP3 has been selected as an attractive candidate gene for the 6q23 region. The product of TNFAIP3, A20, is a potent anti-inflammatory protein that negatively regulates the transcription factor nuclear factor kappa-beta (NF)-κB responses to tumor necrosis factor (TNF) α toll-like receptor and NOD2 signaling.Citation60 Inappropriate NF-κB activity has been linked with many AIDs and inflammatory diseases.Citation61,Citation62 This hypothesis is further supported by TNFAIP3 knockout mice, which develop severe multi-organ inflammation including inflammation of the joints.Citation63 Interestingly, a recent study has shown TNFAIP3 protein expression in human synovium and in vitro evidence of altered TNFAIP3 transcription by 6q23 intergenic SNPs associated with RA, further supporting its role in the pathogenesis of RA.Citation64
Further GWAS
Since the initial WTCCC GWAS, many additional GWAS have been carried out in RA.Citation55,Citation57,Citation65–Citation68 is a summary of the major RA GWAS from 2007 to present, their study designs, and the significant SNP associations reported in each study.
Table 1 Major independent RA GWAS from 2007 to present
Independent RA GWAS have led to the identification and validation of many novel RA susceptibility loci. The major GWAS detailed in evidence novel associations with TRAF1/C5, TNFAIP3, REL, and CCR6. REL represents a good example of an RA locus identified in independent GWAS not previously implicated by association studies.Citation66 Gregersen et al initially genotyped 278,502 SNPs in 2418 cases and 4504 controls from North America to identify the association of the marker SNP rs13031237 with REL. Subsequently this association was confirmed in an independent replication cohort of 2064 cases and 2882 controls and provided convincing evidence in the combined data set (P = 3.08 × 10−14). The association with rs1031237 which maps to an intron in RELCitation67 has since been confirmed in a UK population of 3962 cases and 3531 controls.Citation69 REL is a strong candidate gene for RA as it encodes c-Rel, which is a member of the NF-κB family of transcription factors that are involved in immune system regulation. c-Rel-deficient mice are resistant to induction of collagen-induced arthritis and have also shown deficiency in Th1 type immune responses. c-Rel has also been implicated in the involvement of CD40 signaling pathways with effects on B cell proliferation and survival.Citation66
As well as identifying novel RA loci, independent GWAS studies have been a useful strategy for the validation of previously reported loci and the implication of other highly suggestive associations, some of which have subsequently been confirmed as RA susceptibility loci, eg, BLK. It is well- documented that larger sample sizes increase the power to maximize the probability of being able to detect novel associations with the small effect sizes expected of complex diseases.Citation70 This has since led to the establishment of larger sample collections and the implementation of larger meta-analysis studies.
Meta-analysis studies
Many novel RA loci have been discovered more recently through meta-analysis studies. Meta-analyses combine data from several studies and increase the power of a study to detect novel associations of modest effect. The association with the CD40 susceptibility locus was discovered through a meta-analysis of WTCCC and US GWAS. This association was validated in a replication study of 3929 cases and 5807 matched controls from eight case-control collections.Citation71 Subsequently, it has also been confirmed in a large UK population.Citation72 Association with CD40 has since been replicated in Graves diseaseCitation73–Citation75 and multiple sclerosis (MS).Citation76 The RA susceptibility variant rs4810485 is located in the second intron of the CD40 gene, with the gene itself encoding a protein that is a member of the TNF receptor super-family (TNFSR5). The protein expressed by CD40 is found on the surface of antigen presenting cells including B cells, dendritic cells, and macrophages.Citation70 It is essential in stimulating a broad range of immune and inflammatory responses in several autoimmune conditions which implicates it as a good candidate gene for RA.
The most recent RA meta-analysis included a total of 12,307 patients and 20,169 controls of European descent. Seven novel RA risk alleles were identified at genome-wide significance following replication in an independent data set of 6768 RA cases and 8806 controls.Citation16 The novel SNPs are located in genes related to immune function including CCR6, IL6ST, IRF5, PXK, RBPJ, and SPRED2. Additionally the study confirmed 24 previously established RA loci and further findings of borderline significance were reported that will require further validation.
Candidate gene studies in the GWAS era
Identification of RA loci previously associated with other AIDs
Another strategy that has proved useful in finding new associations with RA is candidate gene studies based on GWAS findings from RA and other AIDs. A good example of this is the 4q27 region which contains four genes: KIAA1109, Tenr, IL2, and IL21. Originally, this region was first associated with T1D and celiac disease in two GWAS.Citation77,Citation78 Based on these findings, Zhernakova et al went on to investigate this disease locus in RA in a case-control study population and found evidence for association with rs6822844.Citation79 Subsequently, this association has been validated in independent Dutch and UK populations. The 4q27 region has since shown association with ulcerative colitis,Citation80 juvenile idiopathic arthritis,Citation81 psoriatic arthritis,Citation82 and Crohn’s disease,Citation83 further implying that this locus is a general risk factor for multiple AIDs. The IL2 and IL21 genes represent strong candidate genes for this region because the proteins encoded by these genes are cytokines that play a significant role in the development and control of inflammation in RA. Animal models of RA have demonstrated a pathogenic role for both IL-2 and IL-21. Mice deficient in IL-2 develop severe autoimmunity because of defective regulatory T-cell production.Citation84 In DBA/1 mice with collagen-induced arthritis, the administration of IL-21 receptor Fc fusion proteins has led to an improvement in clinical symptoms and histological parameters.Citation85 Despite the potential of both these genes as therapeutic targets in RA susceptibility, it will be difficult to identify the causal polymorphisms in this region due to the high degree of LD.
Pathway-based bioinformatics approaches
Gene relationships across implicated loci (GRAIL) developed by Raychaudhuri et al is a method of prioritizing SNPs to follow up in a candidate gene validation study in order to identify novel associations.Citation86 Raychaudhuri et al selected 370 SNPs from 179 loci that reached P < 0.001 in a previous independent GWAS meta-analysis for investigation by GRAIL. GRAIL initially defines a genomic region in LD with each candidate SNP and then selects all genes within the region. It then uses statistical text mining of the available literature in PubMed to assess and score the relatedness of the implicated loci with genomic regions already known to be previously associated with disease. High GRAIL scores implicated 22 loci with functional connectivity. SNPs representing these candidate loci were then genotyped in an independent study of 8096 cases and 11,822 matched controls.Citation87 Three of these loci, CD28, CD2/CD58, and PRDM1, were convincingly replicated. Limitations of the GRAIL method that should be considered include its use of established knowledge bases, which may bias towards more characterized genes, and its assumption that each region only contains one pathogenic gene. It has however proved to be a productive method of analysis to identify novel susceptibility genes.
The wealth of novel susceptibility loci identified in recent GWAS and meta-analysis studies demonstrates that the GWAS approach is still proving to be hugely successful. The use of a more informed, focused candidate–gene approach alongside bioinformatics in the GWAS era has also proved fruitful. The use of these alternative strategies has in turn led to the discovery, replication, and validation of all 34 RA susceptibility loci to date. The wealth of genetic findings for RA has led to challenges and opportunities to consider that are likely to take focus in the post-GWAS era.
What else has GWAS taught us in RA?
GWAS has been greatly successful in identifying numerous RA risk alleles. Many themes have emerged during the search for these loci including: the well-debated concept of ‘missing heritability’, the possibility of genetically distinct clinical subsets of disease, the existence of ethnic heterogeneity, and the evidence of shared risk loci across multiple AIDs. Each of these topics poses a challenge to the complete understanding of the pathogenesis of RA. Ongoing research in this field is vital to help realize the ultimate goal of establishing a relationship between genotype and phenotype.
Missing heritability
Although GWAS have provided valuable insights into the genetic basis of human disease, the identified associations for RA (with some exceptions) have a modest effect size with odds ratios (OR) <1.5. illustrates the heritability estimates for established RA loci. If we consider that the total sibling recurrence risk ratio (λs) is estimated to be between 5 and 10 for RA then based on λs, the established susceptibility loci at present contribute approximately 33%–47% of total heritability for RA.Citation88 This suggests that it is likely that >50% of the genetic risk to RA remains unknown. The concept of ‘missing heritability’ is one of conflicting opinion. Questions remain as to why so much heritability has been left unexplained by GWAS findings when they capture up to 90% of common genetic variation and what it is therefore likely to be. Many explanations of ‘missing heritability’ have been suggested, including; larger numbers of common SNPs of smaller effect sizes (CDCV), rarer and structural variants which are poorly covered in current genotyping arrays, and the concept that the ‘missing’ heritability can be explained by reevaluating the proportion of genetic variance explained by current associations.
Structural variants including copy number variations (CNVs), insertions and deletions, and copy neutral variation such as translocations and inversions are an abundant source of genetic variation. To date, discovery of structural variation has been limited due to the direct focus on SNPs as mapping tools and probable causal variants. CNVs have gained particular attention as methods have improved to detect them but there has been limited work in determining their potential impact on disease risk. Several associations have been made between CNVs and autoimmunity in humans such as psoriasis,Citation89 systemic lupus erythematosus,Citation90 RA,Citation91 Crohn’s disease,Citation92 and T1D.Citation93 A recent GWAS of CNVs in 16,000 cases of eight common diseases and 3000 common controls was carried out by the WTCCC. The findings from this study suggested that common CNVs that can be typed on current genotyping platforms seemed unlikely to account for a large proportion of ‘missing heritability’.Citation94 Other types of structural variation including inversions and translocations have been implicated in rare Mendelian conditions, but remain largely unexplored in complex traits.
The fraction of genetic risk to common diseases that is attributable to rare variants and structural variants is debatable. The detection of rare variation presents a challenge in current genotyping arrays as they have little power to detect SNPs with MAF < 1%, and structural variation is generally under represented in current databases to study effectively. Until the full extent of this variation can be ascertained, it would be premature to conclude what impact it will have on ‘missing heritability’. The pilot phase of the 1000 genomes project has now been completed which has brought the total number of known SNPs to over 15 million, and structural variants to over 20,000.Citation95 The next 5 years will also see a move towards the availability of affordable whole genome sequencing which will help facilitate detailed analysis of rare and structural variants. This will undoubtedly lead to the further identification and refinement of causal variations and help resolve some of the controversy surrounding the concept of ‘missing heritability’.
Other challenges faced in studying complex disease genetics include the role of gene–gene, G–E, and epigenetic modification of the genome. Established G–E associations exist between smoking and HLA-DRB1 SE alleles, PTPN22, and antibodies to cyclic citrullinated peptides;Citation96 however replication of other findings has been poor. These studies are primarily lacking power to detect effects and face controversy and uncertainty in the best ways to model interaction. G–E interactions specifically lack well-characterized environmental exposure data and would benefit from established consortia with uniform data collection and better assessment of exposure variables.Citation97 In essence, better study design, larger sample sizes, and development of analysis techniques will prove essential before substantial progress is made in tackling these issues.
Clinical subsets of disease
RA is currently defined by a broad range of criteria that has been revised recently by the American College of Rheumatology and European League Against Rheumatism.Citation98 RA exhibits extensive phenotypic heterogeneity especially in the early stages of disease and as such is not recognized as a discrete clinical entity. It has been accepted for a long time that there are likely to be several distinct subtypes of RA that will correlate to different genotypes.Citation99
RA is already classified into two subsets of disease by the presence or absence of anti-citrullinated protein antibodies (ACPA), with two-thirds of patients with early RA being ACPA-positive. ACPA are autoantibodies detected in RA patients and are highly predictive of the future development of RA,Citation100 ACPA-positive disease being associated with a more destructive phenotype with higher rates of joint destruction compared to ACPA-negative disease.Citation101 This supports the notion that different mechanisms may be involved in the development of ACPA-positive and ACPA-negative RA. Indeed, evidence is emerging that ACPA-positive and ACPA-negative disease may well vary in associations to different genetic and environmental risk factors.Citation102
The majority of genetic risk factors identified for RA to date have been found in ACPA-positive cohorts. HLA-DRB1 SE alleles predispose mainly to ACPA-positive RA with a smaller effect on ACPA-negative disease,Citation90 contributing 18% to heritability of ACPA-positive disease compared to only 2.4% in ACPA-negative disease.Citation103 Far fewer genetic risk factors have been associated with ACPA-negative RA, but include IRF5, C-type lectin genes, and a suggestive, nonreplicated association with HLA-DRB1*03.Citation102 The lack of risk factors associated with ACPA-negative RA could be due to increased heterogeneity within this disease subset or due to a lack of studies with large numbers of ACPA-negative patients. The heritability of ACPA-negative RA is similar to that of ACPA-positive RA, indicating that many genetic risk factors remain to be identified for ACPA-negative RA. It should be recognized that there are limitations in using an antibody-based biomarker like ACPA. There is still ambiguity in whether ACPA presentation is a cause or effect of disease and whether differences result from interindividual variation in immune response rather than from the underlying disease process.
One goal arising from GWAS is the possibility to stratify patients further than the simple ACPA classification based on the predominant genetic background or biological pathway that is driving their disease. It has been proposed that treating RA as a quantitative trait would be a better approach to dissect its genetic heterogeneity under the assumption that it is a common disorder affected by multiple underlying genes.Citation104 This idea assumes that phenotypes vary along a continuous gradient and that a disorder such as RA can be considered as the extreme of a quantitative trait. Essentially this implies that using the present case-control design will confound genetic analysis, as controls are diluted with individuals who nearly meet the classification criteria. Refinement of statistical analyses will be important in pursuing the identification of sets of variants that could potentially relate to different underlying disease mechanisms and to the verification of different subtypes of disease. Undoubtedly, this will help refine diagnosis, predictions of outcome and treatment response, and will provide the opportunity to develop selective therapies that are specific for an individual’s phenotype.
Ethnic heterogeneity in RA risk loci
Through GWAS, clear differences have emerged amongst the major ethnic groups with regards to the genes underlying RA. Some genetic polymorphisms are restricted to specific ethnic populations, indicating a presence of genetic heterogeneity in RA, whilst some are common across multiple ethnic groups, eg, TNFAIP3 and STAT4.Citation40 One of the most striking differences seen between different populations is the nonsynonymous coding SNP of the PTPN22 gene (1858C > T). Although widely replicated across European populations, this polymorphism is rarely found in Asian populations,Citation105 indicating that genetic heterogeneity might be explained by the presence of exclusive susceptibility alleles in specific populations. There are also clear differences in the association of the PADI4 gene. PADI4 was the first non-HLA genetic risk factor for RA initially demonstrated in a Japanese population.Citation106 It has since been confirmed through replication in multiple East-Asian populations,Citation107,Citation108 but has exhibited reports of conflicting associations in European populationsCitation30,Citation109–Citation111 despite a comparable allele frequency between the two populations. Risk alleles of the HLA-DRB1 locus have also been shown to vary significantly between ethnic groups. Genetic variants restricted to specific ethnic groups could reflect the result of migration, natural selection, or mutation and could explain the significant differences that have been observed between prevalence rates of disease. Studies have shown a much lower prevalence of RA in developing countries compared to European and American populations.Citation112 Differences in prevalence data could however result from heterogeneity in the distribution of environmental exposures, or age-distribution differences between populations. There are many methodological errors such as different classification criteria for case assessment, reporter bias and access to medical care that could also result in differences between prevalence that are not specific to underlying genetic heterogeneity.Citation40 Populations of European ancestry have dominated the majority of GWAS, which could explain the limited replication of genetic associations in other ethnic populations. Some studies that have reported negative findings are based on relatively small sample sizes, which could indicate inadequate statistical power. Challenges lie in collecting large cohorts of homogenous samples from a variety of different populations. As well as conducting GWAS in a variety of larger well-characterized populations, further dissection of genetic and environmental determinants and their interactions will help clarify genetic heterogeneity between ethnic populations with regards to AIDs.
Shared autoimmune risk loci
Another theme that has emerged in the search for RA susceptibility genes is the concept that some genetic susceptibility variants predispose to multiple AIDs which gives an insight to the possibility of shared genetic pathways. shows a selection of the RA susceptibility risk factors that have shown to be associated in multiple AIDs.Citation37–Citation39,Citation44,Citation113–Citation121,Citation122–Citation147
Table 2 Selection of RA susceptibility loci associated with other AID
Since the application of GWAS the number of loci predisposing to multiple AIDs has increased rapidly. It is evident that many loci predispose to more than one AID. A review of recent genetic studies by Zhernakova et al revealed that shared genetic factors between AIDs indicate the possibility of common etiological pathways.Citation140 This notion is supported by the clustering of AIDs within families, the overlap in clinical manifestations, and observation that the HLA locus is associated with most common AIDs. In most cases the same allele is associated with risk across different diseases, which highlights the likelihood of common underlying mechanisms, eg, STAT4. In some cases, the same allele predisposes to risk in one AID and is protective in another.Citation148 A good example of this would be the IL2RA gene region where allelic heterogeneity is known to exist between MS and T1D. At rs11594656 the minor allele A is associated with susceptibility to MS (OR 1.17) and protection from T1D (OR 0.87).Citation145 Opposing risk effects at a locus suggest that predisposition to related diseases could be controlled by over- or underexpression of genes and genomic elements in biological pathways. Alternatively, there are variants that show association between multiple AIDs, but at a different position within the same locus. Multiple genetic variants are associated differently in TNFAIP3 between SLE and RA. The causal polymorphism for genetic loci post-GWAS is generally poorly understood and it is possible if different SNPs are associated with different diseases that the likely role of the gene product in pathogenesis for each of the diseases will vary.
It appears that although there is much sharing of loci between similar diseases, not all loci overlap with all AIDs. This could prove useful in determining subsets of biological pathways perturbed in different diseases, giving an insight into how far common diseases share underlying susceptibility, and locate genes or pathways that uniquely affect single diseases, perhaps determining which one of the related diseases someone is likely to develop.
Challenges in the post-GWAS era: a long way to go
The ultimate goal of genetic research in any disease is to unravel the connection between genotype and phenotype. With a wealth of established RA genetic loci and many emerging themes, we are in a greater position to understand the pathogenesis of RA but there is still a long way to go. Most GWAS studies have concentrated on finding SNP associations that are associated with disease susceptibility but have rarely invested resources in the functional characterization of the identified risk loci. Whilst there are 34 statistically valid genetic loci associated with RA, the majority of causal variants for these associations are not known, with the current exception of PTPN22. This is because GWAS are based on the principle of LD. In regions of strong LD it becomes difficult to distinguish between the causal variant and the neighboring markers in high LD with the causal variant. There are other challenges that lie in the way of assigning functionality to susceptibility SNPs. Although GWAS are uncovering more risk variants, it is apparent that the majority of associations have modest effect sizes (OR: 1.1–1.5) which implies that the functional effect of these SNPs could be subtle and hard to establish. In addition, risk alleles found in GWAS are often assigned to the most compelling gene candidate within the region based on either its proximity to the marker or the biological plausibility of its likely involvement in the pathogenesis of disease, making disease pathway analysis imprecise.
With such a wealth of genetic associations, it seems sensible to invest greater effort in the functional annotation of the GWAS findings to confidently assign association to genes. Initial progress in the post-GWAS era will be first made from identifying the causal variant/s for each RA locus, and then localizing the genetic signal to identify which target gene/s it may affect. The second stage will be to investigate the biological mechanism of the risk allele on causality of disease. There is no established gold standard approach to resolve these aims. This review is going to focus on the initial stage of functional characterization in refinement and localization of the causal genetic variants, the stage currently occupied by many of the 34 confirmed RA loci. Strategies including fine-mapping and bioinformatics tools are logical starting methods that can be applied to associated regions.
Fine mapping/resequencing
The fine mapping approach aims to narrow a region of association and pinpoint the causal variant/s responsible. It involves genotyping all known SNPs within a region to resolve information about which genes in a region are likely to be responsible. The confirmed RA susceptibility loci already show different trends in their associations. For some loci, it appears that there are distinct single variant effects (PTPN22, REL, CTLA4) and others multiple independent effects or haplotypes that confer risk (TNFAIP3 and STAT4, respectively). Ultimately the aim of fine mapping these established susceptibility regions is to identity the causal SNPs and to refine the associations at these loci further. Park et al concluded from their findings that fine mapping studies of reported loci have so far failed to find novel common variants with larger effect sizes than their tagging SNPs.Citation149 Despite this, TNFAIP3 is a good example of how the fine-mapping approach resolved the existence of three independent effects at the RA susceptibility locus, two conferring susceptibility (rs6920220 and rs5029937) and one conferring protection (rs13207033).Citation58 Although the success of fine mapping appears to be minimal, it is in its infancy and the Wellcome Trust has recently completed a large-scale fine-mapping study called Immunochip. Immunochip is a customized Illumina chip with 195,806 SNPs designed for fine mapping and replication of established loci across a multitude of AIDs including ∼4500 cases from UK RA patients. This is the first systematic and comprehensive application of fine mapping to date and analysis of the data should help refine all current RA genetic associations, identify causal variants, clarify the existence of haplotypes, and confirm single or multiple independent effects at each locus. There are several limitations to the Immunochip project, which must be considered. This custom chip has been designed for use in white European populations and therefore will not help to refine or replicate associations in other ethnic groups. The chip contains all known SNPs to date from the dbSNP database and from the pilot data release of the 1000 genomes project in June 2010 but will miss unknown SNPs and rare variants that have not yet been discovered. It is also thought that the Immunochip will not be 100% accurate in genotyping rare and structural variants as it is a difficult process and it is envisaged that the availability of whole genome sequencing will prove to be more informative for these types of variation.Citation150
The above mentioned limitations of fine mapping can be overcome by resequencing susceptibility loci. Resequencing has not been extensively used to identify causal variants at complex diseases risk regions, because it is much more expensive than fine mapping. However, sequencing costs are currently decreasing, which will make possible the utilization of this approach to establish comprehensive maps of genomic variation at disease loci in the near future.
Following the application of fine mapping and resequencing, regions with a number of putative associated SNPs that cannot be separated any further by strength of genetic association will still exist. After defining the target region with identification of candidate SNP variant/s the next step is to use computational methods for SNP prioritization and to build up evidence for their likely functional impact.
Bioinformatics tools to prioritize SNPs
Bioinformatics tools are a further refinement step for the prioritization of causal SNPs. Many tools exist to enable identification of a candidate for the causal variant by utilizing prediction of functional effects to prioritize SNPs for downstream analysis. One such tool is the ENCyclopedia Of DNA Elements (ENCODE) which is hosted by the University of California Santa Cruz (UCSC). The aim of ENCODE is to find and document all the functional elements that exist in the genome in both coding and noncoding regions. This database essentially gathers its data from wet lab experiments. It includes data from a range of experiments in a variety of tissues and cell types including transcription factor binding sites, chromatin profiling, and histone modification. Data generated from wet lab experiments potentially offer greater evidence of putative function compared with the current predictive algorithms. Such a wealth of data is available through UCSC and although incredibly useful is difficult and time consuming to interrogate. Martin et al have developed a bioinformatics program (ASSIMILATOR) that quickly assimilates a concise summary of experimental data for inputted SNPs from UCSC making it much easier to compare and assess their biological relevance.Citation151 This is a very promising method of building functional evidence to support prioritization of SNPs.
Another step towards functional characterization of loci is to explore associations between candidate SNPs and gene expression. Treating gene expression as a quantitative trait makes it possible to correlate gene transcript levels with SNPs (expression quantitative trait loci [eQTLs]). Gene expression levels have been shown to have a strong heritable component and variation can be due to polymorphisms close to the gene locus (cis) or in a different chromosome (trans). Many genetic variants resulting in phenotypic differences are mediated through changes in gene expression, and correlation between gene expression and DNA polymorphisms can be used to aid the interpretation of genetic association, indicating which particular transcript is most likely to be influenced by the associated SNP. Indeed, several studies have already demonstrated that trait-associated SNPs are more likely to be eQTLs.Citation152,Citation153 Whole genome mRNA expression data are becoming publicly available for a growing number of cell and tissue types enabling resources, such as the Gene Regulators in Disease project, to aid the performance of in silicoeQTL analysis and the correlation of genotype to different transcript levels.Citation154 This is a strategy that can potentially help connect risk variants to their target genes. These are just two of the bioinformatic approaches available to refine the location of causal variants and implicate the leading positional candidate SNPs based on functional evidence. Several molecular methods have also proved useful to further investigate genetic localization of association signals, including Allele Specific Expression and 3C, but are beyond the scope of this review.
The secondary challenge of moving from an associated variant to a mechanism of action that explains disease causality is more substantial and will require many additional resources including validated functional assays, animal models, or in vitro models in which the causal variants can be assessed. This will require a lot more time, patience, and expertise to develop. It is anticipated that in the future the functional underpinning of many GWA tag SNPs can be elucidated. Closing the gap between genotype and phenotype is complicated especially since most known loci are outside protein coding regions and many have modest effects. It is important to remember that the attempt to functionally characterize all disease-associated SNPs is just at the beginning and is very far from proving causality of all disease-associated risk loci.
Conclusion
The GWAS approach has been used extensively to find the common genetic risk factors associated with RA. The results of these scans have been remarkable and strong associations with both known and novel genomic regions have been uncovered, expanding the number of robustly associated RA loci to 34. Larger meta-analysis studies have more recently been employed to increase the power to detect novel associations of smaller effect sizes and to find common associations between related AIDs. Ongoing GWA studies alongside statistical and epidemiological refinement will undoubtedly deliver novel associations and potentially implicate further pathways that could be important in the pathogenesis of RA.
GWAS has uncovered several themes that will undoubtedly contribute valuable knowledge to the complete understanding of the pathogenesis of RA. Heritability estimates have suggested that more than 50% of the genetic risk to RA remains unknown. The concept of ‘missing heritability’ is likely to become much clearer once whole genome sequencing can be implemented on a larger scale and the detection of all common, rare and, structural variants is possible. Larger, well-characterized cohorts of patients from different ethnic backgrounds are necessary to fully resolve the existence of genetic heterogeneity between populations.
Whilst the current genetic discoveries have implicated many important pathways, the largest knowledge gap lies in how the established DNA variations contribute to the pathogenesis of disease. It is apparent that the need to establish causal variants and elucidate their biological role in disease is imperative. It is anticipated that the use of fine-mapping data from the Immunochip project will help refine all the current regions of association with RA to identify the causal variants as well as giving more insight into the concept of shared AID loci. Bioinformatics tools can then be used to prioritize SNPs that can be taken forward into functional studies for validation of biological function. The integration of both genomic and functional data will be necessary to prove the role that implicated pathways and genes play in complex diseases. Identification of novel biological pathways susceptible to pharmacological intervention is possible, which in turn will enable the development of effective preventative and therapeutic agents. In addition the likely stratification of RA patients into more genetically homogenous subgroups could well facilitate prediction of disease progression and response to treatment.
Acknowledgments
The authors thank Jane Worthington for critical reading of the manuscript. KM is funded by Arthritis Research United Kingdom. GO is supported by the European Union (Marie Curie IEF Fellowship PIEF-GA-2009-235662).
Disclosure
The authors report no conflict of interest in this work.
References
- WorthingtonJInvestigating the genetic basis of susceptibility to rheumatoid arthritisJ Autoimmun200525Suppl162016257177
- YoungAKoduriGExtra-articular manifestations and complications of rheumatoid arthritisBest Pract Res Clin Rheumatol200721590792717870035
- ScottDLWolfeFHuizingaTWRheumatoid arthritisLancet201037697461094110820870100
- AlamanosYDrososAAEpidemiology of adult rheumatoid arthritisAutoimmun Rev20054313013615823498
- ReginsterJYThe prevalence and burden of arthritisRheumatology (Oxford)200241Suppl 13612173279
- DeightonCMWalkerDJThe familial nature of rheumatoid arthritisAnn Rheum Dis199150162651994873
- WordsworthPBellJPolygenic susceptibility in rheumatoid arthritisAnn Rheum Dis19915063433462059076
- JarvinenPAhoKTwin studies in rheumatic diseasesSemin Arthritis Rheum199424119287985034
- SilmanAJMacGregorAJThomsonWTwin concordance rates for rheumatoid arthritis: results from a nationwide studyBr J Rheumatol199332109039078402000
- MacGregorAJSniederHRigbyASCharacterizing the quantitative genetic contribution to rheumatoid arthritis using data from twinsArthritis Rheum2000431303710643697
- Van der LindenMPFeitsmaALle CessieSAssociation of a single-nucleotide polymorphism in CD40 with the rate of joint destruction in rheumatoid arthritisArthritis Rheum20096082242224719644859
- StoltPBengtssonCNordmarkBQuantification of the influence of cigarette smoking on rheumatoid arthritis: results from a population based case-control study, using incident casesAnn Rheum Dis200362983584112922955
- TobonGJYouinouPSarauxAThe environment, geo-epidemiology, and autoimmune disease: rheumatoid arthritisAutoimmun Rev201095A288A29219944780
- SymmonsDPEnvironmental factors and the outcome of rheumatoid arthritisBest Pract Res Clin Rheumatol200317571772712915154
- OliverJESilmanAJRisk factors for the development of rheumatoid arthritisScand J Rheumatol200635316917416766362
- StahlEARaychaudhuriSRemmersEFGenome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk lociNat Genet201042650851420453842
- MorganAWRobinsonJIConaghanPGEvaluation of the rheumatoid arthritis susceptibility loci HLA-DRB1, PTPN22, OLIG3/TNFAIP3, STAT4 and TRAF1/C5 in an inception cohortArthritis Res Ther2010122R5720353580
- StastnyPAssociation of the B-cell alloantigen DRw4 with rheumatoid arthritisN Engl J Med197829816869871147420
- GregersenPKSilverJWinchesterRJThe shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritisArthritis Rheum19873011120512132446635
- FernandoMMStevensCRWalshECDefining the role of the MHC in autoimmunity: a review and pooled analysisPLoS Genet200844e100002418437207
- LeeHSLeeATCriswellLASeveral regions in the major histocompatibility complex confer risk for anti-CCP-antibody positive rheumatoid arthritis, independent of the DRB1 locusMol Med2008145–629330018309376
- RaychaudhuriSRecent advances in the genetics of rheumatoid arthritisCurr Opin Rheumatol201022210911820075733
- DingBPadyukovLLundstromEDifferent patterns of associations with anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis in the extended major histocompatibility complex regionArthritis Rheum2009601303819116921
- MichouLCroiseauPPetit-TeixeiraEValidation of the reshaped shared epitope HLA-DRB1 classification in rheumatoid arthritisArthritis Res Ther200683R7916646982
- BottiniNMusumeciLAlonsoAA functional variant of lymphoid tyrosine phosphatase is associated with type I diabetesNat Genet200436433733815004560
- BegovichABCarltonVEHonigbergLAA missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritisAm J Hum Genet200475233033715208781
- FaragoBTalianGCKomlosiKProtein tyrosine phosphatase gene C1858T allele confers risk for rheumatoid arthritis in Hungarian subjectsRheumatol Int200929779379619034456
- KokkonenHJohanssonMInnalaLJidellERantapaa-DahlqvistSThe PTPN22 1858C/T polymorphism is associated with anti-cyclic citrullinated peptide antibody-positive early rheumatoid arthritis in northern SwedenArthritis Res Ther200793R5617553139
- PiererMKaltenhauserSArnoldSAssociation of PTPN22 1858 single-nucleotide polymorphism with rheumatoid arthritis in a German cohort: higher frequency of the risk allele in male compared to female patientsArthritis Res Ther200683R7516635271
- PlengeRMPadyukovLRemmersEFReplication of putative candidate-gene associations with rheumatoid arthritis in >4,000 samples from North America and Sweden: association of susceptibility with PTPN22, CTLA4, and PADI4Am J Hum Genet20057761044106016380915
- SimkinsHMMerrimanMEHightonJAssociation of the PTPN22 locus with rheumatoid arthritis in a New Zealand Caucasian cohortArthritis Rheum20055272222222515986352
- VikenMKOlssonMFlamSTThe PTPN22 promoter polymorphism −1123G > C association cannot be distinguished from the 1858C > T association in a Norwegian rheumatoid arthritis materialTissue Antigens200770319019717661906
- WesolyJHuXThabetMMThe 620W allele is the PTPN22 genetic variant conferring susceptibility to RA in a Dutch populationRheumatology (Oxford)200746461762117135225
- ZhernakovaAEerlighPWijmengaCBarreraPRoepBOKoelemanBPDifferential association of the PTPN22 coding variant with autoimmune diseases in a Dutch populationGenes Immun20056645946115875058
- ChelalaCDuchateletSJoffretMLPTPN22 R620W functional variant in type 1 diabetes and autoimmunity related traitsDiabetes200756252252617259401
- ChungSACriswellLAPTPN22: its role in SLE and autoimmunityAutoimmunity200740858259018075792
- CriswellLAPfeifferKALumRFAnalysis of families in the multiple autoimmune disease genetics consortium (MADGC) collection: the PTPN22 620W allele associates with multiple autoimmune phenotypesAm J Hum Genet200576456157115719322
- HinksAWorthingtonJThomsonWThe association of PTPN22 with rheumatoid arthritis and juvenile idiopathic arthritisRheumatology (Oxford)200645436536816418195
- OrozcoGSanchezEGonzalez-GayMAAssociation of a functional single-nucleotide polymorphism of PTPN22, encoding lymphoid protein phosphatase, with rheumatoid arthritis and systemic lupus erythematosusArthritis Rheum200552121922415641066
- KochiYSuzukiAYamadaRYamamotoKGenetics of rheumatoid arthritis: underlying evidence of ethnic differencesJ Autoimmun2009323–415816219324521
- VangTCongiaMMacisMDAutoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variantNat Genet200537121317131916273109
- KormanBDKastnerDLGregersenPKRemmersEFSTAT4: genetics, mechanisms, and implications for autoimmunityCurr Allergy Asthma Rep20088539840318682104
- LeeHSRemmersEFLeJMKastnerDLBaeSCGregersenPKAssociation of STAT4 with rheumatoid arthritis in the Korean populationMol Med2007139–1045546017932559
- KobayashiSIkariKKanekoHAssociation of STAT4 with susceptibility to rheumatoid arthritis and systemic lupus erythematosus in the Japanese populationArthritis Rheum20085871940194618576330
- MartinezAVaradeJMarquezAAssociation of the STAT4 gene with increased susceptibility for some immune-mediated diseasesArthritis Rheum20085892598260218759272
- ZervouMIMamoulakisDPanierakisCBoumpasDTGoulielmosGNSTAT4: a risk factor for type 1 diabetes?Hum Immunol2008691064765018703106
- WursterALTanakaTGrusbyMJThe biology of Stat4 and Stat6Oncogene200019212577258410851056
- KannanKOrtmannRAKimpelDAnimal models of rheumatoid arthritis and their relevance to human diseasePathophysiology200512316718116171986
- RischNMerikangasKThe future of genetic studies of complex human diseasesScience19962735281151615178801636
- ZhuMZhaoSCandidate gene identification approach: progress and challengesInt J Biol Sci20073742042717998950
- LanderESLintonLMBirrenBInitial sequencing and analysis of the human genomeNature2001409682286092111237011
- International HapMap ConsortiumA haplotype map of the human genomeNature200543770631299132016255080
- ReichDELanderESOn the allelic spectrum of human diseaseTrends Genet200117950251011525833
- PritchardJKCoxNJThe allelic architecture of human disease genes: common disease-common variant … or not?Hum Mol Genet200211202417242312351577
- HindorffLAJunkinsHAHallPNMehtaJPManolioTAA catalog of published genome-wide association studies12122010 Available at: http://www.genome.gov/gwastudies/Accessed April 21, 2011
- Wellcome Trust Case Control ConsortiumGenome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controlsNature2007447714566167817554300
- ThomsonWBartonAKeXRheumatoid arthritis association at 6q23Nat Genet200739121431143317982455
- PlengeRMCotsapasCDaviesLTwo independent alleles at 6q23 associated with risk of rheumatoid arthritisNat Genet200739121477148217982456
- OrozcoGHinksAEyreSCombined effects of three independent SNPs greatly increase the risk estimate for RA at 6q23Hum Mol Genet200918142693269919417005
- CoornaertBCarpentierIBeyaertRA20: central gatekeeper in inflammation and immunityJ Biol Chem2009284138217822119008218
- VereeckeLBeyaertRvanLGThe ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathologyTrends Immunol200930838339119643665
- BrownKDClaudioESiebenlistUThe roles of the classical and alternative nuclear factor-kappaB pathways: potential implications for autoimmunity and rheumatoid arthritisArthritis Res Ther200810421218771589
- LeeEGBooneDLChaiSFailure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient miceScience200028954882350235411009421
- ElsbyLMOrozcoGDentonJWorthingtonJRayDWDonnRPFunctional evaluation of TNFAIP3 (A20) in rheumatoid arthritisClin Exp Rheumatol201028570871420822710
- KochiYOkadaYSuzukiAA regulatory variant in CCR6 is associated with rheumatoid arthritis susceptibilityNat Genet201042651551920453841
- JuliaABallinaJCaneteJDGenome-wide association study of rheumatoid arthritis in the Spanish population: KLF12 as a risk locus for rheumatoid arthritis susceptibilityArthritis Rheum20085882275228618668548
- GregersenPKAmosCILeeATREL, encoding a member of the NF-kappaB family of transcription factors, is a newly defined risk locus for rheumatoid arthritisNat Genet200941782082319503088
- PlengeRMSeielstadMPadyukovLTRAF1-C5 as a risk locus for rheumatoid arthritis – a genomewide studyN Engl J Med2007357121199120917804836
- EyreSHinksAFlynnEConfirmation of association of the REL locus with rheumatoid arthritis susceptibility in the UK populationAnn Rheum Dis20106981572157319945995
- SpencerCCSuZDonnellyPMarchiniJDesigning genome-wide association studies: sample size, power, imputation, and the choice of genotyping chipPLoS Genet200955e100047719492015
- RaychaudhuriSRemmersEFLeeATCommon variants at CD40 and other loci confer risk of rheumatoid arthritisNat Genet200840101216122318794853
- OrozcoGEyreSHinksAAssociation of CD40 with rheumatoid arthritis confirmed in a large UK case-control studyAnn Rheum Dis201069581381619435719
- TomerYConcepcionEGreenbergDAA C/T single-nucleotide polymorphism in the region of the CD40 gene is associated with Graves’ diseaseThyroid200212121129113512593727
- KimTYParkYJHwangJKA C/T polymorphism in the 5′-untranslated region of the CD40 gene is associated with Graves’ disease in KoreansThyroid2003131091992514611700
- MukaiTHiromatsuYFukutaniTA C/T polymorphism in the 5′ untranslated region of the CD40 gene is associated with later onset of Graves’ disease in JapaneseEndocr J200552447147716127217
- Australia and New Zealand Multiple Sclerosis Genetics ConsortiumGenome-wide association study identifies new multiple sclerosis susceptibility loci on chromosomes 12 and 20Nat Genet200941782482819525955
- ToddJAWalkerNMCooperJDRobust associations of four new chromosome regions from genome-wide analyses of type 1 diabetesNat Genet200739785786417554260
- Van HeelDAFrankeLHuntKAA genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21Nat Genet200739782782917558408
- ZhernakovaAAlizadehBZBevovaMNovel association in chromosome 4q27 region with rheumatoid arthritis and confirmation of type 1 diabetes point to a general risk locus for autoimmune diseasesAm J Hum Genet20078161284128817999365
- FestenEAGoyettePScottRGenetic variants in the region harbouring IL2/IL21 associated with ulcerative colitisGut200958679980419201773
- AlbersHMKurreemanFAStoeken-RijsbergenGAssociation of the autoimmunity locus 4q27 with juvenile idiopathic arthritisArthritis Rheum200960390190419248117
- LiuYHelmsCLiaoWA genome-wide association study of psoriasis and psoriatic arthritis identifies new disease lociPLoS Genet200843e100004118369459
- MarquezAOrozcoGMartinezANovel association of the interleukin 2-interleukin 21 region with inflammatory bowel diseaseAm J Gastroenterol200910481968197519471255
- MalekTRThe biology of interleukin-2Annu Rev Immunol20082645347918062768
- YoungDAHegenMMaHLBlockade of the interleukin-21/interleukin-21 receptor pathway ameliorates disease in animal models of rheumatoid arthritisArthritis Rheum20075641152116317393408
- RaychaudhuriSPlengeRMRossinEJIdentifying relationships among genomic disease regions: predicting genes at pathogenic SNP associations and rare deletionsPLoS Genet200956e100053419557189
- RaychaudhuriSThomsonBPRemmersEFGenetic variants at CD28, PRDM1 and CD2/CD58 are associated with rheumatoid arthritis riskNat Genet200941121313131819898481
- OrozcoGBarrettJCZegginiESynthetic associations in the context of genome-wide association scan signalsHum Mol Genet201019R2R137R14420805105
- HuffmeierUBergboerJGBeckerTReplication of LCE3C-LCE3B CNV as a risk factor for psoriasis and analysis of interaction with other genetic risk factorsJ Invest Dermatol2010130497998420016497
- MamtaniMRovinBBreyRCCL3L1 gene-containing segmental duplications and polymorphisms in CCR5 affect risk of systemic lupus erythaematosusAnn Rheum Dis20086781076108317971457
- McKinneyCMerrimanMEChapmanPTEvidence for an influence of chemokine ligand 3-like 1 (CCL3L1) gene copy number on susceptibility to rheumatoid arthritisAnn Rheum Dis200867340941317604289
- McCarrollSAHuettAKuballaPDeletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s diseaseNat Genet20084091107111219165925
- GraysonBLSmithMEThomasJWGenome-wide analysis of copy number variation in type 1 diabetesPLoS One2010511e1539321085585
- CraddockNHurlesMECardinNGenome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controlsNature2010464728971372020360734
- DurbinRMAbecasisGRAltshulerDLA map of human genome variation from population-scale sequencingNature201046773191061107320981092
- MahdiHFisherBAKallbergHSpecific interaction between genotype, smoking and autoimmunity to citrullinated alpha-enolase in the etiology of rheumatoid arthritisNat Genet200941121319132419898480
- ThomasDMethods for investigating gene-environment interactions in candidate pathway and genome-wide association studiesAnnu Rev Public Health201031213620070199
- AletahaDNeogiTSilmanAJ2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiativeArthritis Rheum20106292569258120872595
- WeyandCMKlimiukPAGoronzyJJHeterogeneity of rheumatoid arthritis: from phenotypes to genotypesSpringer Semin Immunopathol1998201–25229836366
- BerglinEPadyukovLSundinUA combination of autoantibodies to cyclic citrullinated peptide (CCP) and HLA-DRB1 locus antigens is strongly associated with future onset of rheumatoid arthritisArthritis Res Ther200464R303R30815225365
- Van derWDHuizingaTWEvery shared epitope allele for itself?Nat Rev Rheumatol20095947747819710670
- PadyukovLSeielstadMOngRTA genome-wide association study suggests contrasting associations in ACPA-positive versus ACPA-negative rheumatoid arthritisAnn Rheum Dis201070225926521156761
- van derWDHouwing-DuistermaatJJToesREQuantitative heritability of anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritisArthritis Rheum200960491692319333951
- PlominRHaworthCMDavisOSCommon disorders are quantitative traitsNat Rev Genet2009101287282819859063
- KochiYSuzukiAYamadaRYamamotoKEthnogenetic heterogeneity of rheumatoid arthritis-implications for pathogenesisNat Rev Rheumatol20106529029520234359
- SuzukiAYamadaRChangXFunctional haplotypes of PADI4, encoding citrullinating enzyme peptidylargininedeiminase 4, are associated with rheumatoid arthritisNat Genet200334439540212833157
- IkariKKuwaharaMNakamuraTAssociation between PADI4 and rheumatoid arthritis: a replication studyArthritis Rheum200552103054305716200584
- KangCPLeeHSJuHChoHKangCBaeSCA functional haplotype of the PADI4 gene associated with increased rheumatoid arthritis susceptibility in KoreansArthritis Rheum2006541909616385500
- BurrMLNaseemHHinksAPADI4 genotype is not associated with rheumatoid arthritis in a large UK Caucasian populationAnn Rheum Dis201069466667019470526
- BartonABowesJEyreSA functional haplotype of the PADI4 gene associated with rheumatoid arthritis in a Japanese population is not associated in a United Kingdom populationArthritis Rheum20045041117112115077293
- IwamotoTIkariKNakamuraTAssociation between PADI4 and rheumatoid arthritis: a meta-analysisRheumatology (Oxford)200645780480716449362
- AlamanosYVoulgariPVDrososAAIncidence and prevalence of rheumatoid arthritis, based on the 1987 American College of Rheumatology criteria: a systematic reviewSemin Arthritis Rheum200636318218817045630
- BrandOGoughSHewardJHLA, CTLA-4 and PTPN22: the shared genetic master-key to autoimmunity?Expert Rev Mol Med2005723115
- DieudePGuedjMWipffJThe PTPN22 620W allele confers susceptibility to systemic sclerosis: findings of a large case-control study of European Caucasians and a meta-analysisArthritis Rheum20085872183218818576360
- VangTMileticAVBottiniNMustelinTProtein tyrosine phosphatase PTPN22 in human autoimmunityAutoimmunity200740645346117729039
- DahaNAKurreemanFAMarquesRBConfirmation of STAT4, IL2/IL21, and CTLA4 polymorphisms in rheumatoid arthritisArthritis Rheum20096051255126019404967
- GlasJSeidererJNagyMEvidence for STAT4 as a common autoimmune gene: rs7574865 is associated with colonic Crohn’s disease and early disease onsetPLoS One201054e1037320454450
- LeeYHWooJHChoiSJJiJDSongGGAssociation between the rs7574865 polymorphism of STAT4 and rheumatoid arthritis: a meta-analysisRheumatol Int201030566166619588142
- MoonCMCheonJHKimSWAssociation of signal transducer and activator of transcription 4 genetic variants with extra-intestinal manifestations in inflammatory bowel diseaseLife Sci20108617–1866166720176035
- PrahaladSHansenSWhitingAVariants in TNFAIP3, STAT4, and C12 or f30 loci associated with multiple autoimmune diseases are also associated with juvenile idiopathic arthritisArthritis Rheum20096072124213019565500
- RemmersEFPlengeRMLeeATSTAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosusN Engl J Med20073571097798617804842
- ZhernakovaAEerlighPBarreraPCTLA4 is differentially associated with autoimmune diseases in the Dutch populationHum Genet20051181586616025348
- ZhaoSXPanCMCaoHMAssociation of the CTLA4 gene with Graves’ disease in the Chinese Han populationPLoS One201053e982120352109
- LesterSDownie-DoyleSRischmuellerMCTLA4 polymorphism and primary Sjogren’s syndromeArthritis Res Ther20079340117559691
- EinarsdottirESoderstromILofgren-BurstromAThe CTLA4 region as a general autoimmunity factor: an extended pedigree provides evidence for synergy with the HLA locus in the etiology of type 1 diabetes mellitus, Hashimoto’s thyroiditis and Graves’ diseaseEur J Hum Genet2003111818412529710
- ChistiakovDATurakulovRICTLA-4 and its role in autoimmune thyroid diseaseJ Mol Endocrinol2003311213612914522
- AyadiHHadjKHRebaiAFaridNRThe genetics of autoimmune thyroid diseaseTrends Endocrinol Metab200415523423915223054
- BanYTomerYGenetic susceptibility in thyroid autoimmunityPediatr Endocrinol Rev200531203216369210
- DengYTsaoBPGenetic susceptibility to systemic lupus erythematosus in the genomic eraNat Rev Rheumatol201061268369221060334
- HolmesSFrieseMASieboldCJonesEYBellJFuggerLMultiple sclerosis: MHC associations and therapeutic implicationsExpert Rev Mol Med200573117
- IwasaKKato-MotozakiYFurukawaYUp-regulation of MHC class I and class II in the skeletal muscles of myasthenia gravisJ Neuroimmunol20102251–217117420546939
- LoiseauPLepageVDjelalFHLA class I and class II are both associated with the genetic predisposition to primary Sjogren syndromeHum Immunol200162772573111423179
- PenderMPGreerJMImmunology of multiple sclerosisCurr Allergy Asthma Rep20077428529217547851
- RiouxJDGoyettePVyseTJMapping of multiple susceptibility variants within the MHC region for 7 immune-mediated diseasesProc Natl Acad Sci U S A200910644186801868519846760
- ThomasGPBrownMAGenetics and genomics of ankylosing spondylitisImmunol Rev2010233116218020192999
- WatermanMXuWStempakJMDistinct and overlapping genetic loci in crohn’s disease and ulcerative colitis: correlations with pathogenesisInflamm Bowel Dis20101210 Epub ahead of print
- DieudePGuedjMWipffJAssociation of the TNFAIP3 rs5029939 variant with systemic sclerosis in the European Caucasian populationAnn Rheum Dis201069111958196420511617
- EyreSHinksABowesJOverlapping genetic susceptibility variants between three autoimmune disorders: rheumatoid arthritis, type 1 diabetes and coeliac diseaseArthritis Res Ther2010125R17520854658
- FungEYSmythDJHowsonJMAnalysis of 17 autoimmune disease-associated variants in type 1 diabetes identifies 6q23/TNFAIP3 as a susceptibility locusGenes Immun200910218819119110536
- ZhernakovaAvan DiemenCCWijmengaCDetecting shared pathogenesis from the shared genetics of immune-related diseasesNat Rev Genet2009101435519092835
- NishimotoKKochiYIkariKAssociation study of TRAF1-C5 polymorphisms with susceptibility to rheumatoid arthritis and systemic lupus erythematosus in JapaneseAnn Rheum Dis201069236837319336421
- KurreemanFAGoulielmosGNAlizadehBZThe TRAF1-C5 region on chromosome 9q33 is associated with multiple autoimmune diseasesAnn Rheum Dis201069469669919433411
- HinksAEyreSKeXOverlap of disease susceptibility loci for rheumatoid arthritis and juvenile idiopathic arthritisAnn Rheum Dis20106961049105319674979
- BehrensEMFinkelTHBradfieldJPAssociation of the TRAF1-C5 locus on chromosome 9 with juvenile idiopathic arthritisArthritis Rheum20085872206220718576341
- MaierLMLoweCECooperJIL2RA genetic heterogeneity in multiple sclerosis and type 1 diabetes susceptibility and soluble interleukin-2 receptor productionPLoS Genet200951e100032219119414
- BrandOJLoweCEHewardJMAssociation of the interleukin-2 receptor alpha (IL-2R alpha)/CD25 gene region with Graves’ disease using a multilocus test and tag SNPsClin Endocrinol (Oxf)200766450851217371467
- Blanco-KellyFMatesanzFAlcinaACD40: novel association with Crohn’s disease and replication in multiple sclerosis susceptibilityPLoS One201057e1152020634952
- WangKBaldassanoRZhangHComparative genetic analysis of inflammatory bowel disease and type 1 diabetes implicates multiple loci with opposite effectsHum Mol Genet201019102059206720176734
- ParkJHWacholderSGailMHEstimation of effect size distribution from genome-wide association studies and implications for future discoveriesNat Genet201042757057520562874
- CortesABrownMAPromise and pitfalls of the ImmunochipArthritis Res Ther201113110121345260
- MartinPBartonAEyreSASSIMILATOR: a new tool to inform selection of associated genetic variants for functional studiesBioinformatics201127114414621177990
- NicolaeDLGamazonEZhangWDuanSDolanMECoxNJTrait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWASPLoS Genet201064e100088820369019
- MoffattMFKabeschMLiangLGenetic variants regulating ORMDL3 expression contribute to the risk of childhood asthmaNature2007448715247047317611496
- GeBPokholokDKKwanTGlobal patterns of cis variation in human cells revealed by high-density allelic expression analysisNat Genet200941111216122219838192