Abstract
Congenital nystagmus is a pathologic oculomotor state appearing at about three to four months of age. The precise diagnosis requires detailed clinical examination and electrophysiological findings. This case report presents two male patients with congenital nystagmus examined longitudinally from the age of six months until 17–18 years of age. Clinical and electrophysiological protocols were detailed. The first results showed electronegative electroretinography in the two cases and examination combined with electroretinographic findings helped us to make the diagnosis of Congenital Night Stationary Blindness (CSNB). This diagnosis was confirmed by genetic studies. CSNB is interesting to study because through electrophysiological findings, it enables a better understanding of the physiology of neural transmission in the outer part of the retina.
Introduction
Congenital nystagmus is a pathologic oculomotor state observed before the maturation of visual function, and appearing at about 3–4 months of age. Morphologically, the form may be pure jerk, pure pendular, or both jerk and pendular in different gaze directions. As described by Gottlob,Citation1 nystagmus is a nonspecific manifestation of a wide variety of disorders of the eye, visual pathways, or central nervous system. After cycloplegic refraction, the ophthalmologist will search for a sensory defect, such as cataract, corneal opacities, aniridia, chorioretinal coloboma, and albinism, which are the most frequent causes of such visual abnormalities. Moreover, neurological impairment must be eliminated. With other causes, such as Leber’s amaurosis, achromatopsia, or congenital night stationary blindness (CSNB), the eyes are grossly normal. Clinical analysis of the nystagmus may give clues to the diagnosis. The pendular type of nystagmus is associated with ocular albinism, retinoschisis, achromatopsia, and CSNB.Citation2 These congenital disorders require differentiation by detailed clinical examination, and by electrophysiological findings under photopic and scotopic conditions.
Case reports
Two male patients aged 18 years (Case 1) and 17 years (Case 2) were examined longitudinally. The first examination was performed at six months of age, the next at 10 and 11 years of age, and the last at 17 and 18 years of age.
At six months, we performed refractive measurement, slit lamp examination, funduscopy, and electroretinogram (ERG) studies using the Biophysic Medical Pantops PC2 (v 2.53 FRA 1991; Biophysic Medical, Clermont-Ferrand, France). We used contact lens electrodes (Dencott, Paris, France) for recording. After dark-adaptation for 10 minutes, we recorded a scotopic blue ERG. The patients were then light-adapted for three minutes at 100 cd/m2 and a photopic white and red ERG was recorded.
At 10 and 11 years of age, we performed refractive measurements, funduscopic examination, a Roth 28-Hue color vision test, a Goldman visual field test, and ERG with a Monitor Win5000H (Metrovision, Paris, France). We recorded a photopic red, white, and flicker ERG and a scotopic blue ERG.
At 17 and 18 years of age, we performed refractive measurement, fundi examination, Roth 28-Hue color vision test, Goldman visual field examination, and optical coherence tomography of the macula. To perform ERG studies, we used the Metrovision vision monitor. The ERG and the electro-oculogram (EOG) were recorded as described by the most recent International Society for Clinical Electrophysiology of Vision standard.Citation3
Full-field (Ganzfeld) stimulation was used. Recording electrodes comprised corneal contact lens electrodes. The corneal surface was protected by a nonirritating ionic conductive solution (methylcellulose 0.5%). Topical anesthesia was used. The impedance of the applied electrode measured about 3 kOhms. Reference electrodes were placed near each orbital rim as a reference for the corresponding eye. Ground electrodes were placed on the forehead. The skin was prepared by cleaning, and a conductive paste was used to ensure a good electrical connection. Pupils were maximally dilated with tropicamide 1%.
Electrophysiology protocol
To record a dark trough for the EOG, the room light was turned off and EOG values were recorded for 15 minutes in darkness. The minimum amplitude during this period was designated as the dark trough. The light stimulus was turned on and the EOG was recorded, until the light peak occurred and the signal amplitudes had clearly started to descend, at which point the recording was stopped. When the pupils were dilated, the stimulus intensity was 80 cd/m2.
For rod ERG recording, the patients were dark-adapted for at least 20 minutes beforehand. The stimulus was a dim white flash (0.01 cd/s/m2). The interval between two flashes was 1944 msec (approximately two seconds). We recorded eight accumulations for each patient.
The mixed ERG was produced by a white single flash stimulus (3 cd/s/m2) to the dark-adapted eye. The interval between stimuli was 7222 msec (approximately seven seconds). We recorded eight accumulations for each patient.
Dark-adapted oscillatory potentials were generally obtained from the dark-adapted eye using the same white single flash (3.0 cd/s/m2) and a high-pass filter (100 Hz).
For cone (light-adapted) ERG recordings, the patients were light-adapted for 10 minutes. The stimulus was a white single flash (500 cd/s/m2), and the rods were suppressed by a background with a luminance of 30 cd/s/m2. The interval between stimuli was 500 msec. We recorded 30 accumulations for each patient. A 30 Hz light-adapted flicker ERG was also used to represent the cone system. A white stimulus (500 cd/m2) was used, with approximately 30 stimuli per second.
Results
Examination and recordings at six months of age
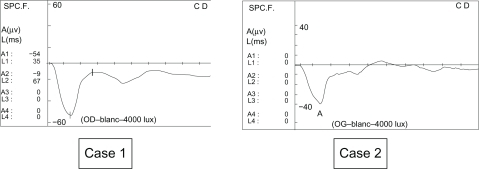
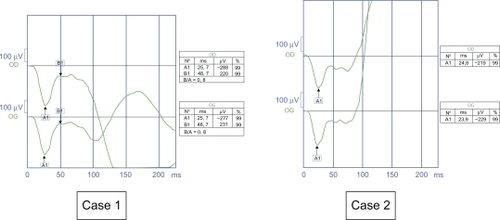
In both cases, nystagmus had been noticed by the mothers at 3–4 months of age. In Case 1, at examination, the nystagmus was a vertical pendular type. In Case 2, the nystagmus was pendular with a horizontal and vertical gaze. Funduscopy and slit lamp examination was normal in both cases. The first ERG at six months, performed under general anesthesia, showed a negative type in our two young patients ().
Figure 1 Negative electroretinogram at six months (1991 and 1992). Case 1. White photopic electroretinogram of the right eye. Case 2. White photopic electroretinogram of the left eye.
Examination and recordings at 10–11 years
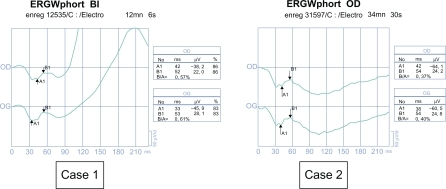
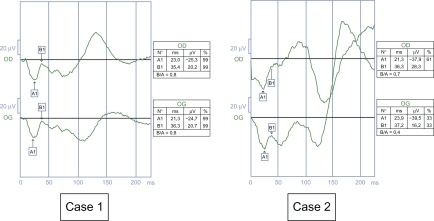
In Case 1, visual acuity was 0.16, with a hyperopic error of +1. The nystagmus was amplified on monocular vision. We observed an esotropia of 12 prism diopters (PD) in the left eye. The fundi stayed normal. In Case 2, visual acuity was 0.1 with a myopic error of 4. Nystagmus was idem on monocular or binocular vision. We did not observe any strabismus. The fundi stayed normal. 28-Hue color vision and Goldmann visual field tests were normal in both cases. The ERG showed a negative profile ().
Figure 2 Electroretinograms stayed negative when they were controlled in 2002 (Case 1, 11 years; Case 2, 10 years). Case 1. Negative profile of the photopic electroretinogram with white flash (b-wave amplitude) 37.8 V in the right eye and 43.1 V in the right eye). Case 2. Negative profile of the photopic electroretinogram with white flash (b-wave amplitude 24.2 V in the right eye and 24.8 V in the left eye).
Examination and recordings at 17–18 years
At the age of 17, Case 1 had a corrected visual acuity of 0.32 with a mild hyperopic refractive error (+1) and showed an esotropia of 16 PD. At the age of 18 years, Case 2 had a corrected visual acuity of 0.2 with a mild myopic refractive error (−4). The fundi remained normal in both cases. Optical coherence tomography of the macula showed no sign of x-linked retinoschisis in either case. Goldman visual field and 28-Hue color vision tests were normal in both cases. This completed our electrophysiological examination.
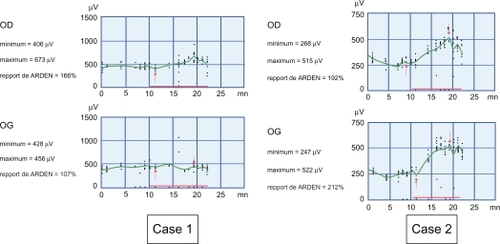
reflects the activity of the retinal pigment epithelium and photoreceptors. In Case 1, it was subnormal in the right eye but affected in the left eye, probably because of the esotropia. In Case 2, it was normal in both eyes. Therefore, the visual pigments and photoreceptors were normal, indicating that the basic abnormality could be in neural transmission at a some level in the outer part of the retina.
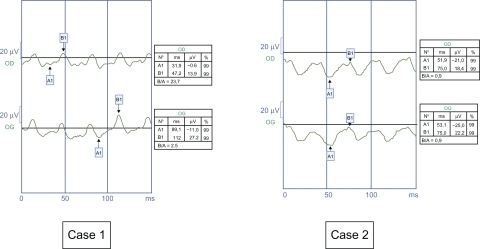
Figure 3 Red arrows indicate the dark trough (first arrow) and the light peak (second arrow) and the red line along the x axis indicates the duration that the background light is on. Case 1. Altered electro-oculogram in the left eye because of esotropia, and subnormal electro-oculogram with Arden ratio 66% on the right eye, probably due to the nystagmus being raised on monocular vision. Case 2. Both eyes are normal (Arden ratio 192% and 212%). The normal value of the Arden ratio is over 185%.
Rod response (dark-adapted) ERG was recordable but lowered (see ). This reflected activity of bipolar cells in the rods. In the complete form of CSNB, the rod response is nonrecordable but is recordable in the incomplete form, albeit lower than in normal subjects.Citation5
Figure 4 Lowered rod response recordable.
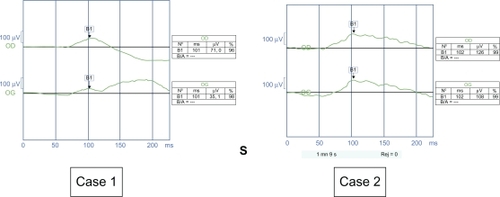
Mixed response (dark-adapted) ERGs showed a negative profile (see ), reflecting impairment of the ON pathway of both rods and cones.Citation4 There was no difference between the complete and incomplete form of CSNB.Citation5
Figure 5 Negative profile of the mixed response.
The cone response (light-adapted ERG) also showed a negative profile (see ) and reflects the impairment of the ON and OFF pathway of the cones.Citation6 Moreover, Miyake et alCitation5 showed that analysis of differences in the b-wave amplitude is very interesting. Indeed, the mean and two standard deviations of the b-wave were 73 ± 25 μV in normal subjects, 52 ± 34 μV in patients with complete CSNB, and 18 ± 26 μV in patients with incomplete CSNB.
Figure 6 Case 1. Negative profile and b-wave amplitude is 20.2 μV on the right eye, 20.7 μV in the left eye, in the range of those of incomplete congenital night stationary blindness. Case 2. Negative profile and b wave amplitude is 16.2 μV and on the left eye, and 32.1 μV on the right eye but in the range of complete congenital night stationary blindness.
The amplitude of the light-adapted flicker response (peak to trough) was smaller and reflected impairment of the cone response by ON or OFF bipolar cells ().Citation3 The mean amplitude and two standard deviations were 43 ± 20 μV in normal subjects, 33 ± 18 μV in patients with complete CSNB, and 11 ± 8 μV in patients with incomplete CSNB.Citation2
Figure 7 Case 1. Amplitude of the flicker response is 12.3 μV in the right eye and 16.2 in the left eye. Case 2. Amplitude of the flicker response is 2.6 μV in the right eye and 2.8 μV in the left eye. These results are compatible with incomplete congenital night stationary blindness.
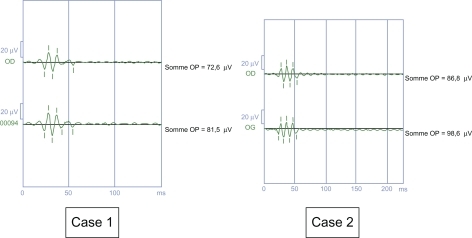
Dark-adapted oscillatory potentials () were lowered, and the smallest was phot-OP 4. Impairment of phot-OP 4 is specific for incomplete CSNB and reflects specific impairment of the cone OFF pathway. In the complete form, phot-OP 2 and 3 are lowered, but phot-OP 4 is normal.Citation5
Figure 8 Reduction of oscillatory potentials, the lowest being phot-OP 4.
Discussion
The basic underlying retinal pathology for all electronegative ERGs is a disturbance at or proximal to the inner segments of the photoreceptors, but relative sparing of photoreceptors and function of the outer segments. The age of onset, type of nystagmus, and the first negative ERG profile led us to the suspicion of an inherited retinal disorder in our patients, in whom we found stationary inherited retinal disease with negative ERG and normal fundi, normal macular optical coherence tomography, normal color vision, and normal visual fields. As a result of the electrophysiological findings, our main hypothesis was CNSB in its incomplete form, because in the complete form of CSNB rod ERG is not recordable, but is recordable and with smaller cone ERG amplitude in the incomplete form. The flicker response is smaller in the incomplete form than in the complete form. Furthermore, in the oscillatory potential, phot-OP 4 is not recordable in the incomplete form whereas it is normal in the complete form.
CSNB was first described in 1952 by Schubert-Bornschein. Clinically, it is an association between an x-linked recessive hereditary mode, a lowered visual acuity, a nystagmus with normal fundi, and a negative ERG. However, there are some differences between patients with CSNB, and a subtype was apparent. Miyake, in 1986, defined this subtype as the incomplete form of CSNB on the basis of electrophysiological findings.Citation7
Pathophysiological studies suggest that patients with complete CSNB have an almost complete block of synaptic transmission from photoreceptors to the ON bipolar cells in both rod and cone visual pathways, whereas the OFF pathway appears to be intact. In contrast, patients with incomplete CSNB have an incomplete defect of the synapses on the ON and OFF bipolar cells in the rod and cone visual pathways.
Evidence of genetic heterogeneity in x-linked complete and incomplete CSNB was reported by Boycott et al in 1998. The gene for incomplete CSNB was identified by Storn et al and Bech-Hansen et al as the pore-forming subunit of a Type 2 voltage-gated calcium channel found in the retina. Loss of this functional channel impairs calcium influx into the rods and cones that is needed for sustaining tonic release of neurotransmitters from the presynaptic terminals.Citation8 The gene for complete CSNB was identified in 2000 by Bech-Hansen as NYX, encoding the extracellular protein, nyctalopin, that guides and promotes the formation and function of the ON pathway of the retina. Therefore, in 2002, MiyakeCitation7 renamed the two types, ie, Type 1 (the complete form) and Type 2 (the incomplete form). Sieving et al analyzed the ERG abnormalities created in anesthetized nonhuman primates by intravitreal application of glutamate analogs that selectively suppress retinal ON or OFF bipolar cell activity.Citation9 2-amino-4-phosphonobutyric acid, which selectively blocks light responses in the ON pathway and depolarizes bipolar cells, fully reproduced the essential ERG abnormalities found in human Type 1 CSNB when the OFF pathway remained active.
Conclusion
These two cases of congenital nystagmus required electrophysiological investigation to make the diagnosis, which were subsequently confirmed by genetic studies. They were shown to be two cases of Type 2 (incomplete) CSNB. It is an important diagnosis to be able to give to young patients, because CSNB is a static disease, unlike retinitis pigmentosa or cone-rod dystrophy, for example, which are degenerative disorders. CSNB is a very interesting area of research because, via electrophysiological findings, it enables a better understanding of the physiology of neural transmission in the outer retina.
Disclosure
The authors report no conflicts of interest in this work.
References
- GottlobINystagmusCurr Opin Ophthalm200011330335
- PiehCSimonsz-TothBGottlobINystagmus characteristics in congenital stationary night blindness (CSNB)Br J Ophthalmol20089223624018227204
- MarmorMFFultonABHolderGEMiyakeYBrigellMBachMInternational Society for Clinical Electrophysiology of Vision. ISCEV Standard for full-field clinical electroretinography (2008 update)Doc Ophthalmol2009118697719030905
- RigaudièreFLe GargassonJ-FOeil et Physiologie de la Vision: V-Exploration électrophysiologique Available from: http://lodel.irevues.inist.fr/oeiletphysiologiedelavision/index.php?id=115. Accessed on February 5, 2011.
- MiyakeYYagasakiKHoriguchiMKawaseYKandaTCongenital night stationary blindness with negative electroretinogram: A new classificationArch Ophthalmol1986104101310203488053
- FultonABHansenRMStimulus-response functions for the scotopic b-waveHeckenlivelyJRArdenGBHandbook of Clinical Electrophysiology of Vision2nd edCambridge, MAThe MIT Press2006
- MiyakeYEstablishment of the concept of new clinical entities – complete and incomplete form of congenital stationary night blindnessNippon Ganka Gakkai Zasshi200210673775512610835
- ManserghFMutation of the calcium channel gene CACNA1f disrupts calcium signaling, synaptic transmission and cellular organization in mouse retinaHum Mol Genet2005143035304616155113
- KhanNWKondoMHiriyannaKTJamisonJABushRASievingPAPrimate retinal signaling pathways: Suppressing ON-pathway activity in monkey with glutamates analogues mimics Human CSNB1-NYX congenital night blindnessJ Neurophysiol20059348149215331616