Abstract
Purpose
To report a case of acute quinine poisoning, document acute and chronic macular changes with optical coherence tomography imaging and fluorescein angiography (FA), and to review the literature on ocular toxicity of quinine.
Methods
A 32-year-old white female presented to our Emergency Department after ingesting over 7.5 g of quinine. She underwent a complete ophthalmologic examination, fluorescein angiography, Stratus time-domain optical coherence tomography (OCT), and electroretinography at 72 hours and 15 months postingestion. Stratus time-domain and Cirrus spectral-domain OCT, fundus autofluorescence, and FA were obtained at 28 months postingestion.
Results
Fluorescein angiography at 72 hours postingestion revealed normal filling times and vasculature. OCT showed marked thickening of the inner retina bilaterally. At 15 and 28 months follow-up, fundus photography and fluorescein angiography demonstrated optic nerve pallor, severely attenuated retinal vessels while OCT showed inner retinal atrophy. Fundus autofluorescence did not reveal any retinal pigmentary abnormalities.
Conclusions
Quinine toxicity as seen by OCT reveals increased thickness with inner retinal hyperreflectivity acutely with development of significant retinal atrophy in the long-term. Fundus autofluorescence reveals an intact retinal pigment epithelial layer at 28 months. These findings suggest that quinine poisoning may produce a direct toxic effect on the inner retina in the acute phase resulting in long-term retinal atrophy.
Introduction
Acute quinine poisoning is a devastating ocular event that can result in severe loss of vision.Citation1 Quinine is an antimalarial agent that is often used for the treatment of restless legs syndrome. The drug has a narrow therapeutic range and carries the risk of multiple side effects such as cinchonism, hypoglycemia, arrhythmias, hemolysis, gastrointestinal intolerance, fever, and nephritis.Citation2 In early 2007, the US Food and Drug Administration restricted the use of quinine solely to the treatment of malaria. Quinine has long been known to be toxic to the eye even at therapeutic levels, causing symptoms such as loss of vision, reduced color vision, and reduced visual fields.Citation3,Citation4 Most visual symptoms occur after a quinine dose greater than 4 g and death can occur with doses exceeding 8 g.Citation5
We describe a patient with profound visual loss after acute quinine poisoning. At 72 hours postingestion, ERG, Stratus time-domain optical coherence tomography (OCT), fundus photography, and fluorescein angiography (FA) were performed on the patient. At 15 months postingestion, time-domain OCT and FA were again performed on our patient. At the 28 months postingestion visit, Cirrus spectral-domain and Stratus time-domain OCT, FA, and fundus autofluorescence (FAF) were performed. To the best of our knowledge, this is the first documentation in the literature of the retinal changes in acute quinine toxicity as seen by OCT and FAF.
Case report
A 32-year-old white female presented to our Emergency Department five hours after attempting suicide by quinine overdose. The dose was estimated to be between 7.5 and 10 g. On initial examination the patient was complaining of tinnitus, nausea, vomiting, as well as total loss of vision. Serum sodium and potassium were low at 133 mEq/L and 3.6 mEq/L, respectively, and the patient’s ECG showed a prolonged QT interval. The patient was admitted to the medical intensive care unit of the Ohio State University Medical Center for monitoring and support.
Ophthalmology was consulted the following morning. The first ophthalmologic exam was performed at the bedside 23 hours postingestion and showed that the patient had no light perception in both eyes. The pupils were fixed and dilated at 8 mm. Fundoscopy revealed optic nerves with normal color and crisp margins, retinal vessels with normal course and caliber, and maculas with diffuse retinal thickening and bilateral cherry-red spots (CRS). Peripheral retinal examination was unremarkable OU. The patient received hyperbaric oxygen therapy per the recommendation of the medical intensive care unit team.
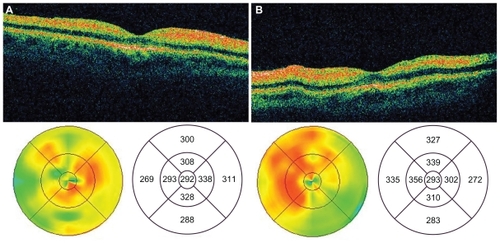
At 48 hours postingestion, the patient reported return of peripheral vision. Examination revealed partial resolution of the CRS with persistent retinal thickening. At 72 hours postingestion, the patient was medically stable enough to be transferred to the ophthalmology clinic. Fundoscopy revealed partial resolution of the CRS (). Fluorescein angiography showed normal filling times and no evidence of leakage or vascular staining (). Stratus time-domain OCT revealed inner retinal hyperreflectivity with thickening in both eyes (). Central subfield thicknesses were 292 microns OD and 293 OS and macular volumes for a 6 mm diagonal were 8.43 mm3 OD and 8.75 mm3 OS. At this visit, bright flash ERG yielded reduced a-waves and absent b-waves, consistent with previous reports on the electrophysiologic effects of quinine poisoning.Citation6
Figure 1 Color fundoscopic images of right a) and left b) eyes 72 hours postingestion demonstrating retinal pallor and cherry red spots. Fluorescein angiograms of right c) and left d) eyes 72 hours postingestion revealing normal retinal vessels.
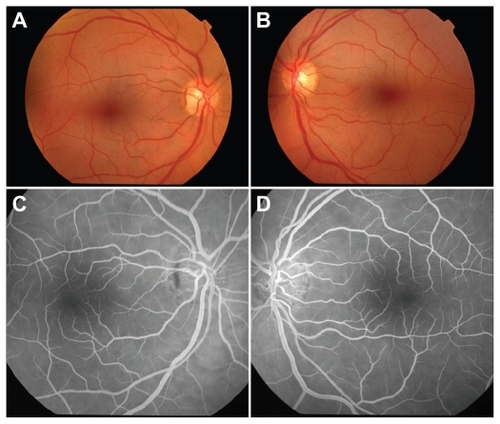
Figure 2 OCT images of the macula of the right a) and left b) eyes 72 hours postingestion demonstrating marked thickening and hyper-reflectivity of the inner retina.
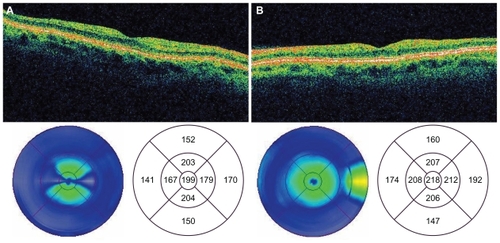
The patient was subsequently lost to follow-up for several months, as she was repeatedly hospitalized in psychiatric institutions and underwent drug rehabilitation. Her first ophthalmologic follow-up examination was performed 15 months postingestion. At this visit, the patient’s uncorrected visual acuity (VA) was 20/20 OU. Although Zeiss-Humphrey 30-2 visual fields demonstrated normal foveal thresholds OU (33 dB OD, 35 dB OS), they were constricted to 15 degrees OU. Dilated fundus examination revealed optic nerve pallor OU with retinal atrophy and severely attenuated retinal arterioles and venules (). However, normal arteriovenous transit times and arteriolar perfusion were demonstrated by fluorescein angiography (). Stratus time-domain OCT revealed severe atrophy of the neuroretina in both eyes with central subfield thickness being 199 microns OD and 218 microns OS. Macular volumes for a 6 mm diagonal had decreased to 4.68 mm3 OD and 5.06 mm3 OS ().
Figure 3 Color fundoscopic images of right a) and left b) eyes 28 months postingestion showing attenuated retinal vessels and optic nerve pallor. FA of the right c) and left d) eyes reveals attenuated vessels without evidence of perfusion deficits.
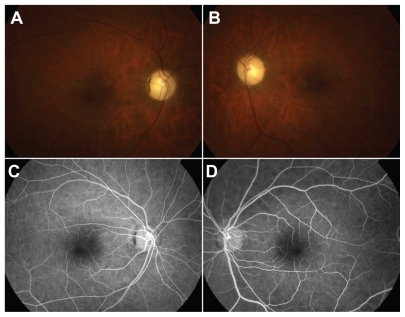
Figure 4 Time domain OCT images of the macula of the right a) and left b) eyes 28 months postingestion showing atrophy of the neuroretina.
The patient was last seen at the Ophthalmology clinic at 28 months postingestion. At this time, her uncorrected VA was 20/20 OD and 20/20 OS. Farnworth D-15 color vision testing demonstrated mild blue-yellow color deficiencies. There were no changes worth noting between the patient’s Humphrey visual fields or FA between this visit and her visit at 15 months postingestion. Cirrus spectral-domain OCT revealed central subfield thicknesses that were 283 microns OD and 279 microns OS and average retinal thicknesses that were 213 microns OD and 211 microns OS (normal average retinal thickness is 264 ± 21.9 microns).Citation7 Although the central retinal thicknesses on the Cirrus OCT were within the normal range, the average macular thicknesses were decreased, a finding consistent with retinal atrophy and to the peripheral constriction seen on the visual fields. FAF photography (TRC-50DX; Topcon, Oakland, NJ) revealed a generalized hyperfluorescent pattern without evidence of atrophic retinal pigment epithelial changes (, ).
Figure 5 Spectral OCT images of the right a) and left b) eyes 28 months postingestion showing atrophy of the neuroretina.
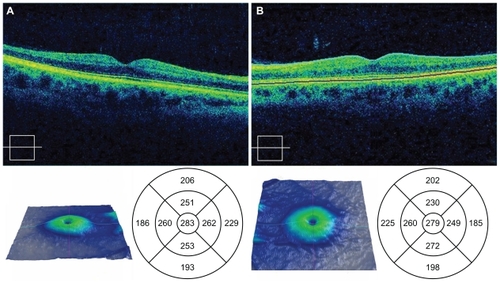
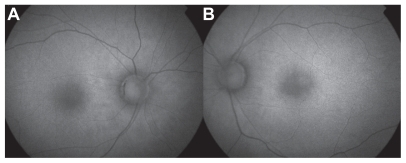
Figure 6 Fundus autofluorescence of right a) and left b) eyes 28 months postingestion demonstrating diffuse hyperfluorescence without evidence of RPE atrophic lesions OU.
Discussion
The presentation and ophthalmologic clinical findings of this patient appear typical of quinine-induced ocular damage. Previous case reports have demonstrated that patients usually develop loss of vision between 10 and 24 hours after ingestion of toxic levels of quinine. Most patients experience a return of central acuity with restricted visual fields over the next several weeks to months. Vessel attenuation and disk pallor seen at our patient’s 15- and 28-month examinations are consistent with the typical late fundus findings of quinine toxicity.Citation5
Despite the compound’s well-established ocular toxicity, the mechanism behind the damage it mediates is not entirely known. Towards this end, the acute OCT and FA findings in our patient were enlightening. At 72 hours postingestion, there was marked inner retinal hyperreflectivity with retinal thickening. This finding is consistent with OCT performed on the ischemic retina during central retinal artery occlusion (CRAO).Citation8 However, in our patient the FA in the acute stage demonstrated no abnormalities in arteriovenous transit and retinal perfusion. FAF at 28 months did not reveal any retinal pigment epithelium atrophic changes. Cirrus spectral-domain OCT demonstrates an intact external limiting membrane as well as inner and outer segment of the photoreceptor layer at 28 months. These findings suggest that quinine poisoning produces damage that is confined to the inner retinal layers.
The presence of a CRS during the acute phase of quinine poisoning in our patient is consistent with previous reports of quinine overdose. CRS formation is seen with ischemia of the ganglion cell layer (GCL) in CRAO and in conditions where storage materials or metabolites accumulate in the GCL of the retina such as congenital gangliosidosis (Tay Sachs disease and Sandhoff’s disease), sphingolipidosis (Niemann-Pick’s disease), glucocerebrosidosis (Gaucher’s disease), and with patients on long-term parenteral nutrition.Citation9,Citation10 In addition to quinine, CRS has also been observed with toxic interactions following intravitreal aminoglycoside injection, ingestion of dapsone, and ingestion of methanol.Citation11–Citation13 In a histopathologic report on primates, Conway et al reported that high doses of intravitreal gentamicin led to retinal toxicity with the formation of a CRS. The retinal toxicity was primarily confined to the inner retina with relatively intact photoreceptor and retinal pigment epithelial layers. In the acute phase, there was nerve fiber and ganglion cell swelling with subsequent atrophy of these layers in the chronic phase. They postulated that retinal toxicity was primarily due to a direct neurotoxic effect on the inner retina.Citation14 This mechanism appears to be most consistent with the clinical, OCT, and FAF findings in our patient. The optic nerves do not appear to be affected in the early stages of toxicity as their appearance was unremarkable and there was no papillary leakage or staining on FA at 72 hours. In the latter stages of follow-up the pattern of visual field loss was one of generalized constriction rather than a cecocentral scotoma, a pattern more consistent with diffuse retinal rather than optic nerve toxicity.
Quinine has long been known to be neurologically active. Quinine is a Class IA anti-arrhythmic that blocks sodium and potassium channels in cardiac myocytes, causing a delay in the initiation of action potentials and increasing the action potential duration.Citation15 Various studies have shown quinine capable of blocking voltage-gated outward potassium channels, stimulating intracellular inositol-1,4,5-triphosphate production, and activating intracellular pathways via guanine nucleotide binding protein (G-protein)-coupled receptors.Citation16–Citation18 These pharmacologic actions of quinine could be responsible for the clinical observations of rapid onset bilateral vision loss and eventual neuronal damage. Prolonged depolarization of the cell membrane can lead to depolarization of the mitochondrial membrane and stimulation of mitochondrial-mediated apoptosis.Citation19,Citation20
Several proposed mechanisms of direct quinine-retina interaction exist in the literature. Aloisi et al postulated that retina damage is a result of free radicals generated by the photoionization of quinine.Citation21 It has long been suspected that quinine blocks acetylcholine transmission in the retina. Recent evidence shows quinine to have an antagonistic effect on α9α10 nicotinic acetylcholine receptors elsewhere in the body.Citation22 Acetylcholine blockade can explain the immediate total blindness observed in quinine overdose, but does not explain how quinine leads to eventual cell death. Perhaps the most promising research concerning the mechanism of quinine toxicity involves gap junctions and connexins. Gap junctions are formed by structural proteins known as connexins and allow for intracellular electrical communication by linking the cytosol of two consecutive cells and are found in gastrointestinal smooth muscle, cardiac muscle, and retinal cells. These tissue types are all affected in quinine poisoning. Studies in transfected mammalian cells have shown that quinine has the ability to act as a reversible, concentration-dependent gap junction channel blocker.Citation23 Furthermore, quinine has been shown to block specific connexins, including connexin 36 (Cx36), which has been found in mouse and rat retinas.Citation24,Citation25 Cx36 gap junctions are responsible for excitatory and inhibitory interneuron communications between retinal ganglion cells and amacrine cells.Citation26 The interaction of quinine with these junctions may help explain the acute blindness seen in quinine toxicity and may also provide greater understanding of the mechanism of quinine’s neurotoxic effect.
Our clinical experience and recent literature support the theory that quinine is directly neurotoxic to the inner retina. More research is needed to identify the exact mechanism by which quinine mediates direct neuronal cell death. Interventions geared towards disrupting quinine’s toxic neuronal activity would perhaps be able to prevent the devastating loss of vision associated with this agent.
Disclosure
No conflicts of interest were declared in relation to this paper.
References
- BrintonGSNortonEWZahnJRKnightonRWOcular quinine toxicityAm J Ophthalmol1980904034107425057
- BarrocasAMCymetTCinchonism in a patient taking quinine for leg crampsComp Ther200733162163
- VusirikalaBWilliamsTARamARA late presentation of ocular quinine toxicity managed with a combination of vasodilatory treatmentsEye20051980180215359264
- BaligaRSWingoCSQuinine induced HUS-TTP: an unusual presentationAm J Med Sci200332637838014671503
- BolandMERoperSMHenryJAComplications of quinine poisoningLancet198513843852857431
- VerdonWClinical electrophysiology in quinine induced retinal toxicityOptom Vis Sci200885172618174833
- KiernanDFHariprasadSMChinEKKiernanCLRagoJMielerWFProspective comparison of Cirrus and Stratus optical coherence tomography for quantifying retinal thicknessAm J Ophthal2007147226727518929353
- ArashvandKCentral retinal arterial occlusionN Engl J Med200735684117314343
- VintonNEHeckenlivelyJRLaidawSAVisual function in patients undergoing long-term total parenteral nutritionAm J Clin Nutr1990528959022122712
- SuvarnaJCHajelaSACherry-red spotJ Postgrad Med200854545718296811
- BrownGCEagleRCShakinEPGruberMArbizioVVRetinal toxicity of intravitreal gentamicinArch Ophthalmol1990108174017442256847
- AbhayambikaKChackoAMahadevanKNajeebOMPeripheral neuropathy and haemolytic anaemia with cherry red spot on macula in dapsone poisoningJ Assoc Physicians India1990385645652174032
- MckellarMJHidajatRRElderMJAcute ocular methanol toxicity: clinical and electrophysiological featuresAust NZ J Ophthalomol199725225230
- ConwayBPTabatabayCACampochiaroPAD’AmicoDJHanninenLAGentamicin toxicity in the primate retinaArch Ophthalmol19891071071122910268
- HillRJDuffHJSheldonRSDeterminants of stereospecific binding of type I antiarrhythmic drugs to cardiac sodium channelsMol Pharmacol1988346596632848186
- CummingsTAKinnamonSCApical K+ channels in Necturus taste cells. Modulation by intracellular factors and taste stimuliJ Gen Physiol1992995916131597680
- SpielmanAINagaiHSunavalaGDassoMRapid kinetics of second messenger production in bitter tasteAm J Physiol1996270C926C9318638676
- MingDRuiz-AvilaLMargolskeeRFCharacterization and solubilization of bitter-responsive receptors that couple to gustducinProc Natl Acad Sci U S A199895893389389671782
- FerreiroEOliveiraCRPereiraCMThe release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathwayNeurobiol Dis20083033134218420416
- LevrautJIwaseHShaoZHVanden HoekTLSchumackerPTCell death during ischemia: relationship to mitochondrial depolarization and ROS generationAm J Physiol Heart Circ Physiol2003284H549H55812388276
- AloisiGGBarbafinaACantonMDall’AcquaFPhotophysical and photobiological behavior of antimalarial drugs in aqueous solutionsPhotochem Photobiol20047924825815115297
- BallesteroJAPlazasPVKracunSEffects of quinina, quinidine, and chloroquine on α9α10 nicotinic cholinergic receptorsMol Pharmacol20056882282915955868
- SrinivasMHopperstadMGSprayDCQuinine blocks specific gap junction channel subtypesProc Natl Acad Sci U S A200198109421094711535816
- SöhlGGüldenagelMTraubOWilleckeKConnexin expression in the retinaBrain Res Brain Res Rev20003213814510751663
- HidakaSKatoTMiyachiEExpression of gap junction connexin36 in adult rat retinal ganglion cellsJ Integr Neurosci2002132215011262
- SchubertTDegenJWilleckeKHormuzdiSGMonyerHWeilerRConnexin36 mediates gap junctional coupling of alpha-ganglion cells in mouse retinaJ Comp Neurol200548519120115791644